
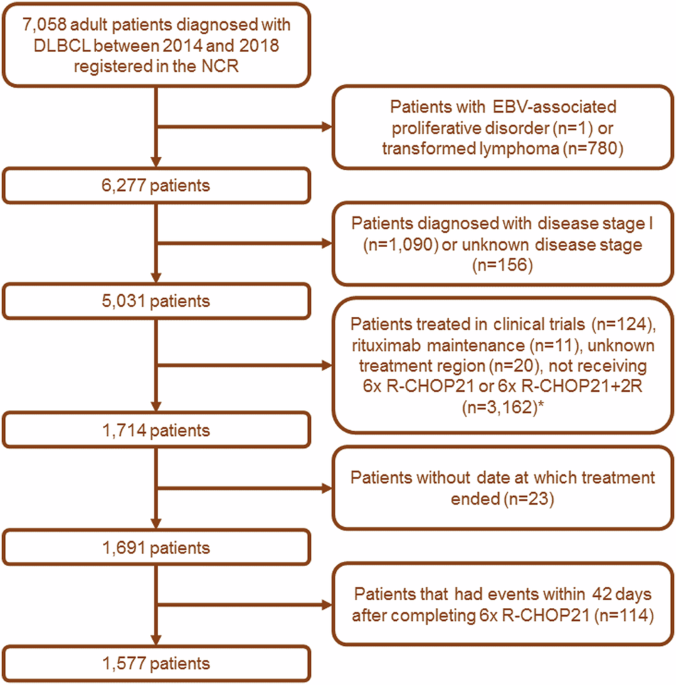
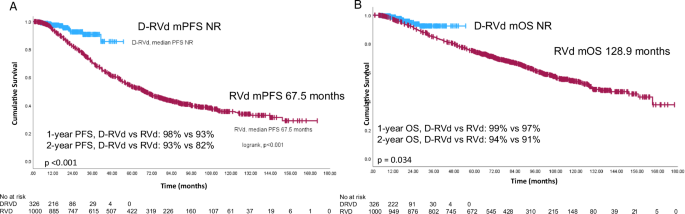
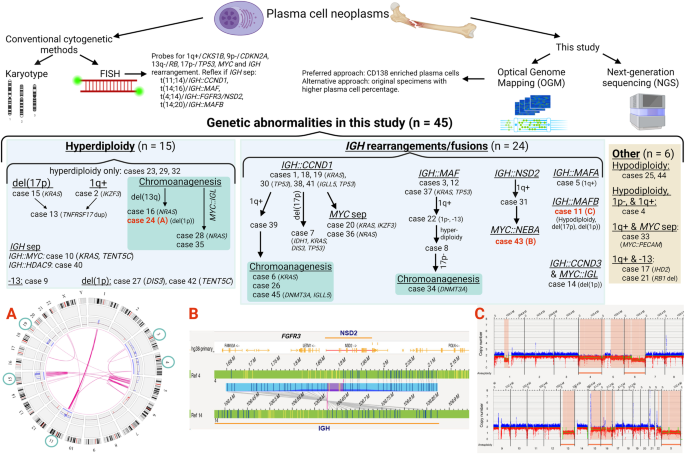
Remember me







CoMMpass IA19 RNA-Seq and CNA data were available for 659 patients. The mean age in the dataset was 62.5 ± 10.7 years; 60% were male, and the ISS distribution was 35% stage I, 35% stage II, and 30% stage III. For the cohort, the 5-year PFS rate was ~32%, with the longest survival time listed at 8 years. An overview is presented in Supplementary Table 1.
Hierarchical clustering using Ollivier-Ricci curvature differentiates subtypes with low progression-free survival ratesThe largest connected network component from shared information between the HPRD, RNA-Seq, and CNA data consisted of 8468 nodes and 33,695 edges. ORC, a correlate for robustness of strength between gene interaction pairs, was computed for each of the 33,695 interaction pairs in each individual patient. Hierarchical clustering of the resultant ORC matrix together with CNA data produced 8 clusters (Supplementary Fig. 1A, Fig. 3A), while clustering based on RNA-Seq produced 6 clusters (Supplementary Fig. 1B, Fig. 3B); both methods being significant for PFS (CNA; p = 0.0082, RNA-Seq; p = 0.0016, log-rank test). Interestingly, the clustering appears to be defining biological differences not captured by the ISS prognostic score, with a relatively even distribution of ISS stages in each cluster.
Fig. 3: Hierarchical clustering using Ollivier Ricci Curvature (ORC) predicts progression-free survival (PFS) in multiple myeloma.
Kaplan–Meier analysis of PFS based on ORC according to (A) copy number aberration, and (B) RNA sequencing. To better understand the differences between the high risk and low risk cohorts, clusters with similar outcomes were grouped. C For CNA based clustering, clusters 1–6 and 8 were combined into the low-risk group. Cluster 7 was the high-risk group. D For RNA-sequencing data, clusters 4 and 6 were combined into a high-risk group. Clusters 1 and 3 were combined into a low-risk group.
Considering the dominant impact of hyperdiploidy on CNA analyses, we repeated hierarchical clustering on the non-hyperdiploid samples and found PFS prediction remained significant (p = 0.0002, log-rank test). Of note, analyzing CNA via ORC produced a cluster representing 10% of patients with a markedly inferior PFS when compared to the remaining clusters (Fig. 3A, C); median PFS was 1.7 years, despite only 35% of patients being ISS III. When assessing previously described copy number risk factors (Supplementary Table 2), patients in this cluster almost universally contain aberration in chr1q (gain; 57%, amplification; 29%, diploid 3%), while also harboring the highest proportion of the complex structural variant chromothripsis (43% of patients, p < 0.0001 compared with the remaining clusters, Fisher’s exact test). This finding is congruent with previously published data demonstrating chromothripsis to be an independent prognostic factor in MM [23], and with an increasing body of knowledge demonstrating that multiple genomic insults compound to worse survival [23, 33].
Clustering of the ORC matrix with RNA-Seq data produced more variation in PFS between clusters (Fig. 3B, D). Of note, clusters 2 and 3 contain the majority of t(11;14) patients (Supplementary Table 3). Considering the dominant role of CCND1 in MM pathophysiology, we repeated hierarchical clustering in the non-t(11;14) samples, which remained significant for PFS-prediction (p = 0.0002, log-rank test). When clustering with all patients; 98% of those in cluster 4 harbor t(4;14), and 81% of those in cluster 6 have a translocation affecting MAF,MAFA or MAFB, with 72% having increased APOBEC-mutational activity. Clusters 1 and 5 are more heterogenous, with a combination of hyperdiploidy, canonical translocations, gain/amp1q, TP53 aberration and chromothripsis. While a high proportion of patients in the 2 clusters with the shortest PFS (4 and 6) carry a previously described genomic risk factor, the other clusters (1 and 3) demonstrate a longer PFS despite 29.2% being ISS III, and 34% harboring a risk factor included in R-ISS / R2-ISS. Given that clustering with ORC using RNA-Seq demonstrated better discrimination of PFS compared with CNA, we have elected to focus on RNA-Seq for the remainder of the current study. A heatmap showing the subject distribution between the two clustering results is shown in Supplementary Fig. 2. Heatmaps showing both the distribution of common markers of MM by both cluster label and patients are presented in Supplementary Fig. 3 for CNA based clustering and Supplementary Fig. 4 for RNA-seq based clustering. We hypothesized that expanding on the ORC analysis with gene set enrichment analysis (GSEA), prognostic modeling, and network topology analysis will provide further biological insights.
Expression analysis using ORC-based risk groups demonstrates differential DNA damage and immune system signalingDifferential gene expression analysis was conducted comparing high-risk (clusters 4 and 6) and low-risk (clusters 1 and 3) as defined by ORC analysis of RNA-Seq data. Gene sets enriched in the high-risk group includes inflammatory response, IL-6/JAK/STAT3 signaling and DNA damage response (DDR) signaling (P53 pathway, DNA repair and apoptosis, Table 1). Of note, there was no significant difference between the groups in p53 function by traditional methods (TP53 mutations and del17p), therefore our methods are capturing more global dysregulation in DNA damage signaling than is evident by standard mutation and copy number analysis.
Table 1 Differential gene expression analysis according to ORC-based risk groups.Within these differentially expressed pathways, 118 genes were selected for further pathway analysis (having absolute log fold change >3.5 and corrected p-value < 0.05, Supplementary Table 4). Of these 118 genes, 19 were under-expressed and 99 were overexpressed in the short survival group compared to the longer survival group in the poor survival group. Of these 118 genes, the majority of them are “bridge genes,” with the rest being hub genes and genes which only form a singular connection with another gene. There were 23 genes that formed a single connection, 80 bridge genes that formed connections with 2–16 genes, and 15 genes which formed >16 connections—which is twice the average number of connections in the HPRD network. Furthermore, of the 118 genes, none overlapped with the MM gene list presented in [34] and only WEE1 was in common with the gene list GEP70 [13]. Compared to the subjects identified by GEP70 as high-risk versus this study, there was a 58% overlap, consistent with expectation from a complementary analysis, and demonstrating that our methods are capturing novel biological features.
In univariate analysis, 8/118 genes were predictors of PFS (BUB1, MCM1, NOSTRIN, PAM, RNF115, SNCAIP, SPRR2A and WEE1, Table 2), with 5 of these also being significant when analyzing based on CNA (NOSTRIN, PAM, RNF115, SNCAIP and SPRR2A). We note a gene dosage effect for RNF115 related to chr1q copy number gain. Average RNA-seq values by cluster are reported in Supplementary Table 5 and a plot showing the relationship between RNA-seq and CNA for RNF115, a 1q gene, is shown in Supplementary Fig. 5. Interestingly, none of these 8 genes feature in previously described lists of MM driver genes [33, 35], suggesting that we are capturing novel aspects of MM biology. In addition to differential expression in the inflammatory response and IL-6/JAK/STAT3 signaling gene sets, interrogation of the ImmuneSigDB database demonstrated 110 /118 genes to overlap with ImmuneSigDB pathways, including all 8 of the independently prognostic genes (Table 2). Taken together, these findings suggest that global assessment of gene interactions can detect complex immune dysregulation.
Table 2 Gene expression in 8 novel immune-network genes associate with survival.Local neighborhood 1-hop and 2-hop gene networks demonstrate differential DNA damage and immune system signalingA key feature of gene network analysis is the ability to capture a wide range of gene-pair interactions, above and beyond the expression levels of a single gene. While this analysis may be difficult to interpret in the context of highly connected genes, it can detect complex patterns (i.e., an overall increase or decrease in network robustness) or specific individual interactions (i.e., a gene-pair demonstrating an increase in robustness while all other local connections become more fragile).
Comparing high-risk and low-risk clusters as defined by ORC analysis of RNA-Seq data, we note several interesting network expression patterns. Within DDR-signaling, TP53 and ATM signaling pathways overwhelming become more robust in the high-risk group (Fig. 4A, B), with more robust pathways generally expected to exert increased effects. While we typically associate loss of p53 function with poor prognosis in cancer, global network analysis is detecting global changes in expression that may not fully capture functional protein levels. The same analysis performed on the basis of CNA demonstrates a mixture of TP53 connections becoming more robust and more fragile, possibly reflecting the impact of del17p (Supplementary Fig. 6A).
Fig. 4: Local neighborhood of selected genes relevant to MM biology and the immune system.
Each line or edge represents the interaction between a gene-pair in a network, comparing the median interactions observed in the high-risk group compared with those in the low-risk group. Blue edges indicate that the connections are more robust in the high-risk group, while orange edges are more fragile, risk being defined by the RNA-Seq-based clustering analysis. A: TP53, B: ATM, C: CCND1, D: MYC, E: IL6, F: IFNGR1, G: TNFRSF17, H: CD38, I: IKZF3. Higher resolution images are available at www.github.com/aksimhal/mm-orc-subtypes.
In addition to DDR-signaling, networks centered on CCND1 and MYC become more robust overall (Fig. 4C, D), which suggests these signaling and transcriptional hubs remain dominant in the context of high-risk disease. In contrast to the above networks showing a clear signal of robustness, the effect on RAF / RAS / MAPK and NFKB signaling are more heterogenous (Supplementary Fig. 6B–D), suggesting that some parts of this network may play an oversized role in MM biology compared with the other interactions.
Considering the immune dysregulation observed on GSEA analysis, signaling through some cytokines and receptors become more fragile (i.e., IL-6, IFNg; Fig. 4E, F), while others demonstrate a more heterogenous effect (i.e., TNF, IFNa; Supplementary Fig. 6F, G). In this context, pathways becoming more fragile would be expected to exert less than normal control. Interestingly, multiple networks involving therapeutic targets for MM immune-based therapies become more fragile, suggesting potential therapeutic vulnerabilities. This included TNFRSF17 (encoding for BCMA, a cellular-therapy target), CD38 (the target of monoclonal antibody daratumumab), IZKF3 (a target of immunomodulatory agent lenalidomide) and SLAMF7 (the target of monoclonal antibody elotuzumab) (Fig. 4G-I, Supplementary Fig. 6H, I).
From the list of 8 novel genes having expression associated with high-risk MM, all have a recognized role in immune regulation (Table 2). In contrast with the other genes, only WEE1, (encoding for a tyrosine kinase which affects G2-M transition), has been previously implicated in MM biology [36]. In the HPRD, WEE1 acts as a hub gene, forming an above average number of connections with its immediate neighbors (18 versus 8.4 for the whole graph). Interestingly, within the 8 prognostic genes, BUB1 and WEE1 connect to each other in a 2-hop analysis via PLK1, CDK1, and CRK. From the genes with significantly different expression between risk groups, 24/118 (20.3%) connect to the 8 prognostic genes within the two-hop analysis.
The 8 genes identified play different roles in their local neighborhoods (Fig. 5); NOSTRIN, (a nitric oxide synthase trafficker), RNF115, (an E3 ubiquitin ligase), and SPRR2A (induced by type-2 cytokines in response to infection) form bridge-like connections to a single other gene. NOSTRIN connects to another nitric oxide gene, NOS3, RNF115 to the RAS oncogene family member RAB7A, while SPRR2A connects with EVPL (associated with squamous cell cancer and autoimmune disease). Four genes act as bridges for their local neighborhood: BUB1, MCM6, PAM, and SNCAIP (Figs. 5, 6). While these genes are not hub genes per se, they connect to multiple hub genes and could therefore play a modulating role.
Fig. 5: Local neighborhood of the eight genes identified as being predictive of PFS.
Each line or edge represents the interaction between a gene-pair in a network, comparing the median interactions observed in the high-risk group compared with those in the low-risk group. Blue edges indicate that the connections are more robust in the high-risk group, while orange edges are more fragile, risk being defined by the RNA-Seq-based clustering analysis. A: BUB1, B: MCM6, C: NOSTRIN, D: PAM, E: RNF115, F: SNCAIP, G:SPRR2A, H: WEE1. Higher resolution images are available at www.github.com/aksimhal/mm-orc-subtypes.
Fig. 6: ‘Two-hop’ neighborhood of the eight genes identified as being predictive of PFS.
Each line or edge represents the interaction between a gene-pair in a network, comparing the median interactions observed in the high-risk group compared with those in the low-risk group. Blue edges indicate that the connections are more robust in the high-risk group, while orange edges are more fragile, risk being defined by the RNA-Seq-based clustering analysis. A: BUB1, B: MCM6, C: NOSTRIN, D: PAM, E: RNF115, F: SNCAIP, G: SPRR2A, H: WEE1. Higher resolution images are available at www.github.com/aksimhal/mm-orc-subtypes.
For example, in the 2-hop analysis, the mitotic checkpoint kinase BUB1 connects to HDAC1 (Fig. 6A), a histone deacetylase commonly upregulated in MM cells with a well-defined impact on prognosis [37]. We note multiple network connections between BUB1 and HDAC1, as well as connections between BUB1 and each of CDK1 (cell-cycle transition regulator) and APC (a tumor-suppressor protein within the Wnt signaling pathway). PAM, encoding for a protein with multiple functions described, connects to PRKCA, a protein kinase involved in regulation of proliferation, tumorigenesis, and inflammation. Interestingly, the network connections around PRKCA are predominantly more robust in the high-risk group. SNCAIP, (which inhibits ubiquitin ligase activity), connects with PTN (Fig. 6F; a hub gene encoding for a protein having a role in cell survival, angiogenesis and tumorigenesis), previously noted to be elevated in MM patients [38]. Our analysis finds that the connection between SNCAIP and PTN becomes more robust in the high-risk group. Interestingly, when comparing the 1- and 2-hop networks between RNA-Seq and CNA data, several gene networks were highly analogous between the two methods (Supplementary Fig. 7).
Overall, the complex gene interactions captured through ORC analysis have the capacity to significantly improve our understanding of biological differences between patients have short and long survival, extending on what we understand from traditional mutation and copy number analysis.
Comments (0)