
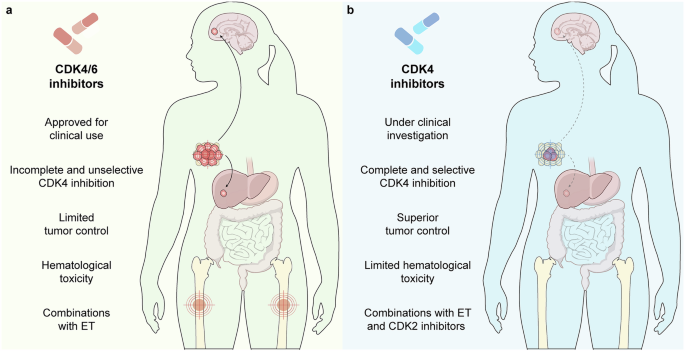
Remember me



In a recent study, Hickey et al. use quantitative proteomics to show that ER-phagy and Golgiphagy are preferentially activated during nutrient stress, and that YIPF3 and YIPF4 function as Golgiphagy receptors.
Macroautophagy (hereby referred to as autophagy) is an important lysosomal degradation mechanism for organelles and macromolecules. Mammalian cells activate autophagy in response to nutrient stress, such as amino acid deficiency, to remodel the proteome according to cellular needs and recycle biosynthetic units.1 This process is often considered non-selective regarding the cellular components that are sequestered by autophagosomes. On the other hand, protein misfolding, damaged membrane-bound organelles, protein aggregation or bacterial infection can stimulate selective autophagy pathways, which rely on autophagy receptors to selectively couple cargo to the autophagosomal membrane.2 Several recent studies have raised the hypothesis that cells induce selective autophagy of the endoplasmic reticulum (ER-phagy) to alleviate nutrient stress.3 Hickey et al.4 address this question using a smart quantitative proteomics strategy and present an extensive profile of autophagic cargoes upon nutrient starvation, revealing that endoplasmic reticulum (ER) and also the Golgi complex are priority targets for cellular recycling. This implicates that ER-phagy and now Golgiphagy play a major role in cell adaptation to nutrient stress. The authors also identified the transmembrane proteins YIPF3 and YIPF4 as bona fide Golgiphagy receptors (Fig. 1).4
Fig. 1: ER- and Golgi-phagy: primary responses to nutrient stress.
Proteomics revealed major organelle proteome remodeling via ER-phagy and Golgiphagy. YIPF3 and YIPF4 act as selective Golgiphagy receptors under starvation and neuron differentiation. mATG8 mammalian ATG8, LIR LC3-interacting region, FIR FIP200-interacting region. Created with BioRender.com.
To uncover “candidate autophagy proteins (CAPs)” related to nutrient starvation, wild-type (WT) and autophagy-deficient cells were treated with starvation medium (EBSS) or amino acid-depleted medium. Using stringent criteria of quantification, complementary proteome analyses discovered ~700 CAPs. To be classified into this cluster, the degradation of the protein upon nutrient starvation must be impaired in autophagy-deficient cells and it must have an abundance profile similar to known autophagy receptors or adaptors. Application of these criteria resulted in a striking enrichment of ER and Golgi proteins among the CAPs, even though they are less abundant in the cell (4.4% and 0.8% of the proteome, respectively). To better understand this effect, the authors considered other factors like non-autophagy-based degradation and translational suppression that affect protein levels during starvation. Based on their proteomic data, the authors calculated the extent to which the protein copy number in each compartment contributed to the overall loss of protein abundance during starvation and discovered that a large part of the reduction in Golgi membrane protein levels was due to selective autophagic degradation of proteins designated as CAPs. The endosome and ER also saw a substantial reduction in protein levels due to degradation of CAPs. The number of ER and Golgi molecules selected for autophagy rivaled that of the much more abundant cytosolic molecules, indicating that ER-phagy and Golgiphagy underlie a substantial contribution to the process.
To study the molecular details regulating these changes, the authors again relied on elegant proteomics approaches that led to the identification of the transmembrane proteins YIPF3 and YIPF4 as Golgiphagy receptor candidates. Using complementary proximity biotinylation assays, YIPF3 and YIPF4 were shown to interact directly with GABARAPL2 and LC3B, two molecules that decorate autophagosomal membranes. It was proposed that YIPF3 and YIPF4 form heterodimers indicating that both proteins work together. YIPF3 and YIPF4 possess disordered N-terminal regions which are oriented to the cytosol and contain functional LC3-interacting regions (LIRs), a feature shared with membrane-embedded ER-phagy receptors, such as FAM134 family members3 and TEX264.5,6
Besides recruiting cargoes, autophagy receptors are typically substrates of the process. To examine whether YIPF3 and YIPF4 fulfill this characteristic, both proteins were fused to the C-terminus of Keima to measure their autophagic flux. Nutrient stress triggered YIPF3/YIPF4 flux in an autophagy-dependent manner. Interestingly, degradation of YIPF3/4 (and TEX264) upon starvation depended on GABARAPs, but not LC3 family proteins. This evidence suggests that GABARAPs could participate in Golgiphagy.
The authors also visualized the mobilization of YIPF3/4 in response to nutrient starvation using fluorescence microscopy. Co-treatment with EBSS and Bafilomycin A1 (an inhibitor of autophagosome-lysosome fusion used to stabilize autophagosomes) resulted in co-localization of YIPF4 with LC3B and LAMP1, suggesting that YIPF4 is transported from the Golgi to the lysosome via autophagosomes. ATG9 vesicles derived from the trans-Golgi network were not affected in YIPF3/4-knockout (KO) cells. This indicates that autophagosome biogenesis is not impaired in the absence of YIPF3/4. Also, CALCOCO1, previously reported as a regulator of Golgiphagy,7 was not involved in the YIPF3/4-mediated process. It would be expected that Golgiphagy affects Golgi structure and function. In the fluorescence microscopy experiments, YIPF3/4 deletion did not alter Golgi morphology; however, ultrastructural studies could provide further details on this aspect. There are several open questions: How are Golgi ribbons and stacks affected? Besides nutrient stress, is Golgiphagy activated during mitosis, when the stacks disassemble into small vesicles? Is the intra-Golgi protein transport and glycosylation regulated by YIPF3/4-mediated autophagy? And is Golgiphagy part of quality control mechanisms?
Further proteomics experiments provided insights into the cellular role of YIPF3/4-driven Golgiphagy. YIPF4-KO cells (therefore also YIPF3) were used to determine cargo selectivity during nutrient stress. Deletion of YIPF4 specifically impaired the degradation of Golgi membrane proteins and showed little effect on Golgi-associated proteins and non-Golgi CAPs, such as ER CAPs. GALNT2 emerged as one of the proteins most strongly stabilized in the absence of YIPF3/4. GALNT2 is involved in O-linked glycosylation and has been functionally linked to the regulation of energy metabolism.
To further expand and confirm the role of YIPF3/4 in Golgi remodeling, the last part of the study was dedicated to investigating whether Golgiphagy is regulated during neuronal differentiation in vitro. Previous findings by this group showed that differentiation of hESCs into induced neurons (iNeurons) is accompanied by an extensive remodeling of the Golgi and ER proteomes, which is dependent on core autophagy machinery.8 In this experimental model, YIPF4 played a specific and essential role in facilitating the degradation of Golgi membrane proteins in iNeurons. Thus, YIPF3 and YIPF4 function as Golgiphagy receptors during not only nutrient starvation but also cell differentiation.
This study revealed that ER-phagy and Golgiphagy operate in parallel to maintain cellular homeostasis under stress and differentiation. Mechanistically, it can be hypothesized that YIPF3/4 clusters induce membrane curvature, similar to FAM134B in ER-phagy.9,10 Because defective ER-phagy has been linked to neurodegeneration, it is tempting to speculate that Golgiphagy is also required for proper neuronal function. Further research will elucidate the molecular mechanisms of Golgiphagy and its influence in cellular physiology and metabolism. This work has already provided the first steps towards achieving this goal.
Comments (0)