
記住我



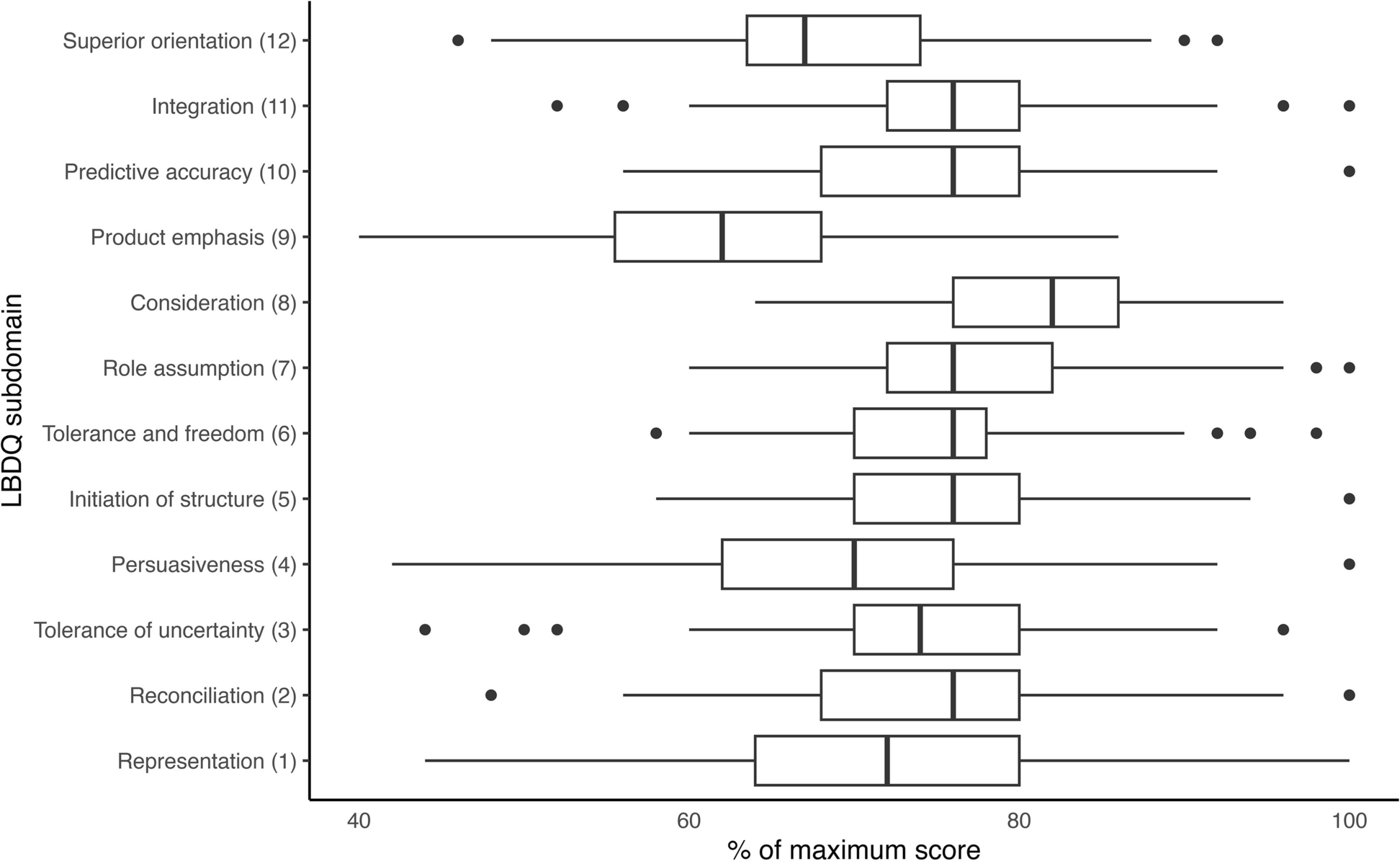


Table 2 presents the timings of when study outcome measures will be collected. Table 3 details the definition and methods to calculate the study outcomes.
Table 2 Schedule of delivery of intervention and data collectionTable 3 Outcome measure definitionsPrimary outcomeEffectiveness of pain relief from randomisation to arrival at hospital as measured by Sum of Pain Intensity Difference (SPID) score (using a 0–10 numerical rating scale(NRS). As this is a pragmatic trial, no fixed time interval was specified to record pain scores, however we did request that pain scores were documented regularly from the point of initial IMP administration to arrival at the hospital.
Secondary outcomesEffectiveness of pain relief and overall patient experience from randomisation to arrival at hospital
Total Pain Relief (TOTPAR) score
Time to perceptible analgesia
Time to meaningful analgesia
Time to peak analgesia
Duration of analgesia
Requirement for rescue analgesia
Proportion of patients with a pain intensity score below 4/10 (0–10 numerical rating scale (NRS)) on arrival at hospital
Vital signs (oxygen saturation, blood pressure, heart rate, respiration rate, Glasgow Coma Scale)
Patient Global Impression of Change on arrival at hospital
Incidence of side effects and adverse events
Airway: vomiting, aspiration, advanced airway management
Respiratory: desaturation, need for ventilatory support
Cardiovascular: arrhythmia, hypotension and hypertension
Neurologic: sedation, excitatory movements, adverse behavioural reactions
Other: nausea, allergic reaction
Resource use
Ambulance job cycle time (scene arrival to arrival at hospital)
Number of ambulance resources (technicians, paramedics, doctors and vehicles) in attendance
Cumulative IMP doses administered
CT scan use
Hospital or ICU admission
Length of stay ED, ICU, Hospital
Longer term outcomes
Chronic pain using Brief pain inventory-short form (BPI-SF) at 3 & 6 months from randomisation
Health-related quality of life measured using the EQ-5D-5L at 3 and 6 months from randomisation
Cost-effectiveness expressed in terms of incremental cost per quality-adjusted life year (QALY) gained
Sample sizeIn line with IMMPACT recommendations, our primary outcome reports Sum of Pain Intensity Difference (SPID). Reductions in pain severity can be reported as either change in NRS, the pain intensity difference (PID), or as a percentage change PID (%PID). The International Association for the Study of Pain has quantified clinically meaningful improvements in pain intensity. Depending on how severe the initial pain is, clinically important improvements in PID range from 1.3 to 5.2. Those with severe pain will need to experience a greater reduction in pain than those with mild pain to experience a clinically important reduction in pain. Similarly, improvements in %PID range from 20.1 to 56.1% depending on severity of pain. Previous studies have established that improvement in PID is equivalent to improvement in SPID [39].
The study has been powered to identify change in SPID calculated using the change in PID. To ensure our study is able to detect at least a 20% improvement in %SPID, regardless of baseline pain intensity, our sample size calculation is powered to detect 20% improvement in %PID, which in turn is equivalent to a 1 point difference (0–10 NRS) in effectiveness between morphine and ketamine.
Previous randomised controlled trials comparing ketamine and morphine have adopted a standard deviation of 3.0 [35, 36, 40, 41]. A review of existing prehospital studies identified that the average non-response/withdrawal rate was 14% [40, 42,43,44,45]. We therefore calculate our sample size assuming a standard deviation of 3.0, 1:1 randomisation, a power of 90%, significance level of 5% and a withdrawal/non-response rate of 15%.
Based on these estimates we calculate our trial will require a sample of 446 subjects, recruiting 223 to each arm of the study, to detect a 1 point difference on the NRS (range 0–10) in primary outcome between morphine and ketamine.
Data analysisThe statistical analysis will follow the estimand framework [46]. Primary analysis will be by intention to treat. The primary outcome will be the SPID, a measure of the area under the curve of the pain score difference from baseline over time. The primary outcome will be analysed using a linear regression. Both unadjusted and adjusted (for important covariates) estimates and 95% confidence intervals for the treatment effect will be obtained. The adjusted estimates will form the basis for the primary analysis. Descriptive summaries of the outcomes will be presented as frequencies or means and medians. For the adjusted estimates, the covariates used will be age (< 60; ≥ 60 years), gender, weight and alternative parenteral IV paracetamol prior to randomisation (as a dichotomy split by yes or no). Age and gender have been chosen as covariates as these groups can experience pain differently, administration of IV paracetamol prior to randomisation is included as a covariate as it is an adjunctive treatment that may impact pain response. Weight is included as a covariate since different weight groups have different requirement for an adequate dose of IMP. For continuous secondary outcomes, analysis will be carried out in a similar way to the primary outcome. For categorical outcomes, logistic regression models will be used. Participant vital signs are recorded in a longitudinal format, for these outcomes we will use a mixed effect model.
Various intercurrent events have been identified for this trial, in line with the estimand framework [46], and approaches to dealing with these have been considered. For discontinuation of the allocated treatment and use of rescue analgesia, we will follow the treatment policy, where the data is analysed as observed. Compliance with administration of the trial drug as per the trial protocol will be monitored. If there is a noticeable degree of non-compliance, we will carry out a complier average casual effects (CACE) analysis [47, 48]. If sufficient data permits, sensitivity analysis will be conducted on a modified intention to treat population, excluding participants that were randomised but the drug was not given. Again, if sufficient data permits, participant deaths will be analysed using Pocock’s win ratio method, this allows death to be interpreted as a participant outcome and infer if the intervention is significantly better than the standard care having considered the clinical priority.
Item missingness is expected for the primary outcome due to the method of recording data used in the trial, however the primary outcome is a measure of the area under a curve, therefore if two pain measurements are recorded the primary outcome can still be measured. Imputation methods have been considered for missingness, but methods such as last observation carried forward are not reliable due to the variability of pain scores, and we cannot assume that pain scores are missing at random therefore multiple imputation is also not feasible. Analyses and template tables will be reported in a detailed statistical analysis plan for review and approval by the Data Monitoring Committee (DMC), prior to final statistical analysis of the data.
Subgroup analysisWe have selected the following subgroups to explore interactions relating to age, gender and the administration of intravenous paracetamol prior to randomisation. The primary outcome will be used as the dependent variable, interaction between the subgroup variable and treatment will be included as an independent variable. Linear regression models will be used to assess the subgroup effect, using interaction terms, subgroup by treatment, to measure the effect of each subgroup.
Data securityParticipant data are being stored on a secure database in accordance with the Data Protection Act (1998). A unique trial identification number is used on all follow-up questionnaires. Warwick clinical trials unit does not receive nor process any personal identifiable data for this trial.
Data collection and managementSource documents are where data are first recorded, and from which participants’ case report form (CRF) data are obtained. These include, but are not limited to, ambulance service records and hospital records (from which secondary outcome data will be collected from). Patient eligibility and ambulance data will be collected from electronic patient records, whereas patient hospital data will be obtained from retrospectively from medical records. Follow up data is obtained from questionnaire packs posted out to participants that have consented to receive them.
On all trial-specific documents, other than the signed consent form, the participant will be referred to by the trial participant number/code, not by name. Data will be entered on to the trial database by the research team.
Health economic evaluationA health economic evaluation has been embedded into the PACKMaN trial. The economic evaluation will take the form of a within-trial cost-effectiveness analysis, conducted from the perspective of the UK NHS and personal social services [49]. Estimates of economic costs will capture resource use associated with the pre-hospital emergency response and broader utilisation of hospital and community-based health and social care services. Resource use in the pre-hospital stage will be extracted from trial case report forms completed by research paramedics. This will include the number of paramedic staff, technicians, doctors, and ambulance staff attending the patient, in addition to transport vehicle, duration of emergency response and cumulative morphine or ketamine doses administered, and medication for treatment of adverse events. Additionally, index admission hospitalisation data resource use will be extracted, this includes length of stay, and number of days receiving critical care and associated critical care level. Resource use questions completed by participants at each assessment point during the study follow-up will provide a profile of all other hospital inpatient and outpatient services, community health and social care encounters, prescribed medications, NHS supplies, time off work and out of pocket medical expenses. Health-related quality of life will be measured using the EQ-5D-5L at three and six months after randomisation. For ethical, logistical, and pragmatic reasons, it is not possible to capture baseline EQ-5D-5L measurements in patients suffering acute pain following trauma within this trial. This is not uncommon within trials in emergency and critical care settings [50]. The baseline analysis for the health economic evaluation will use a fixed baseline approach for EQ-5D-5L health utilities for all participants. This fixed value will be derived by mapping the ‘typical’ acute pain trauma case to the EQ-5D-5L using expert opinion. The sensitivity of this assumption will be tested within sensitivity analyses. Sensitivity analyses will include assigning different values to patients according to severity as determined by registration to the Trauma Audit and Research Network (TARN). TARN can be used as a proxy for severity as the most serious trauma patients will be registered onto TARN whilst less severe cases will not (non-TARN). We will then use expert opinion to estimate a baseline EQ-5D profile for both TARN and non-TARN patients. We will estimate QALY profiles for each participant over a six-month time horizon using the baseline-adjusted area-under-the curve method. We will fit a bivariate regression of costs and QALYs, with multiple imputation of missing data. We will estimate the incremental cost per QALY gained for the comparator interventions from incremental costs and incremental QALYs generated from the regressions. Cost-effectiveness estimates will also be generated for subgroups as specified in the health economics analysis plan.
The primary trial-based analysis will focus on the costs and QALYs accrued during the trial period. There is however potential for costs and benefits to accrue beyond the trial period. If outcomes have not converged by the 6 month timepoint we will consider extrapolating the results over a longer time horizon using a decision analytic model. This would involve combining the trial data with external sources to estimate the long-term cost-effectiveness of the intervention. Any costs and benefits accruing after the first year would be discounted at a rate of 3.5% per year and full probabilistic sensitivity analysis would be conducted in line with the NICE reference case [49]. A decision around the construction of a separate decision analytic model will be made following discussion between the health economists and the trial team following preliminary analysis of the data. This will be informed by considerations such as the conclusiveness and direction of within trial results. For example, if the control dominates the intervention and extrapolation would only increase the strength of this result then there is little need to extrapolate further as the intervention should be rejected. The decision to extrapolate cost-effectiveness will also take into account the availability and quality of external data to inform model parameter inputs.
Adverse eventsThe trial is enrolling patients with acute traumatic injuries which may be immediately life threatening, or result in hospitalisation, persistent or significant disability/incapacity and or death. Potential adverse events are captured on the case report form and investigated by the research paramedic in the first instance. The research paramedic passes the results of their investigation to the PI to determine if the event is an adverse event or not. The trial team are then informed of the outcome as required. In addition, hospital clinicians are able to report clinical concerns to the research paramedic for review that will similarly be reported via the PI to the CI and onward as necessary.
Clinically predictable side effects will be captured but may not be classified as adverse events. For example, morphine is known to cause respiratory depression. A reduction in respiratory rate will be captured on case report forms. If no intervention is required, this will not be classified as an adverse event. However, if the treating paramedic has to assist ventilations of administer naloxone, then this would be classified as an adverse event.
The following adverse events are captured on the case report form as secondary outcomes. If deemed serious they will also be recorded and reported using the SAE form.
Airway: vomiting, aspiration, advanced airway management
Respiratory: desaturation, need for ventilatory support
Cardiovascular: arrhythmia, hypotension and hypertension
Neurologic: sedation, excitatory movements, adverse behavioural reactions
Other: nausea, allergic reaction
Serious adverse events which are not related to the acute traumatic injury, or are complications resulting from the IMP administration to 30 days post trial drug will be reported to the PACKMaN Trial team as soon as possible and within 24 h of the research staff becoming aware of the event.
ReportingResults from the PACKMaN trial will be reported to a trial registry within 12 months of a database lock.
The trial will be reported in accordance with the Consolidated Standards of Reporting Trials (CONSORT) guidelines (Fig. 1) [51].
Fig. 1 Dissemination
DisseminationOur dissemination strategy will target policy makers, commissioners, trauma networks, ambulance services, healthcare providers, academic audiences, patients and the public, charities and advocacy groups. It will include presentations at national and international conferences. We will submit publications to open access peer reviewed journals, develop a lay summary and infographic of the research findings. We will work with our patient and public partners to develop patient stories which effectively communicate key messages from the study. We will publicise via press releases to established media contacts and use our website, blog, Facebook page and Twitter feed to communicate our findings. Our research will support the development of an evidence-based pain management guideline for paramedics by NHS ambulance services. It will improve healthcare quality for patients with severe pain following trauma by engaging clinicians, patients, ambulance services and policy makers to provide better care, by reducing variation in practice and optimising the use of limited health resources.
Data monitoring committeeProfessor Siobhan Creanor (Chair), Professor Julia Williams, Dr Charlotte Small.
Trial steering committeeDr Fionna Moore (Chair), Tim Edwards, Andy Collen, Caroline Leech, Jonathan Bishop, Claire Hulme, Maria Devlin.
CollaboratorsPACKMaN Study Group: chief investigator: Professor Gavin Perkins. Co-chief investigator: Dr Michael Smyth. Co-investigators (Grant holders): Dr Joyce Yeung, Professor Ranjit Lall, Dr Gordon Fuller, Professor Stavros Petrou, Dr Allison Walker, Dr Julian Mark, Duncan Buckley. Senior project manager: Kath Starr. Trial co-ordination/administration: Dr Hannah Noordali. Research fellows/assistants: Felix Michelet (medical statistics), Kamran Khan (Health economics). Patient representative: Duncan Buckley. Trial statistician: Professor Ranjit Lall. Health economist: Professor Stavros Petrou. Intervention development: MODEPHARMA Limited. Data programming team: Ade Willis, Chockalingam Muthiah.
Data sharingThe trial statisticians and DMEC will have access to the dataset for the analysis of trial outcomes. Once the main analyses have been undertaken, deidentified individual participant data will be available to principal and other investigators subject to approval of data analysis plans by the TSC and compliance with the University of Warwick SOPs on Data Management and Sharing. We will comply with Data Sharing Policies that may be instituted by the NIHR during the lifetime of the project.
Ethics approvalWest of Scotland REC 1 approved 01/09/2020.
Provenance and peer reviewThe study was independently peer reviewed as part of the funding application to the NIHR.
留言 (0)