
Remember me
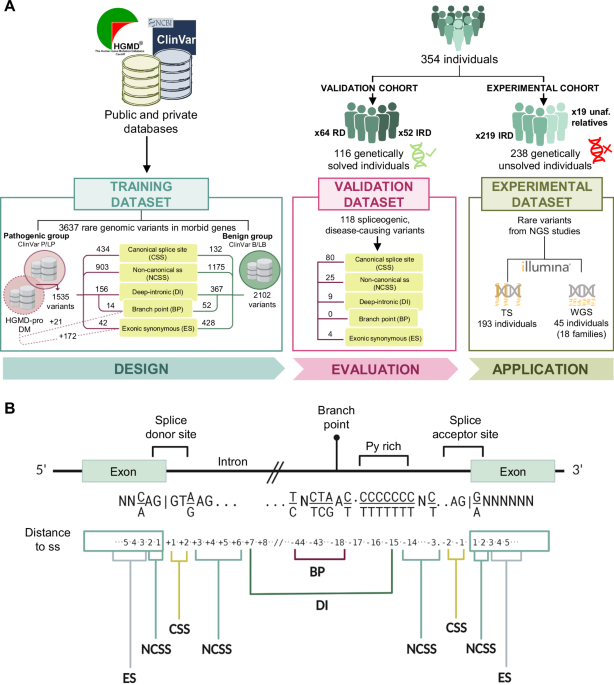


The most common features were mild to moderate short stature, delayed carpal bone ossification and epimetaphyseal dysplasia in the lower limbs. Height ranged from −5,8 to −1,2 SDS. A summary of the clinical observations can be found in Table 1 and Fig. 2, while the radiological findings are summarized in Tables S2, 3, Figs. 3–5 and Figure S2.
Table 1 Summary of clinical features and RPL13 variants.Fig. 2: Pedigrees and clinical pictures of affected individuals.
a Pedigrees of the families. In families 1, 2, and 5, the affected individuals (filled symbols and indicated with +) were heterozygous for c.548G>A in RPL13. Individuals marked with WT were tested and showed the wild-type allele. In family 4, the affected individuals were heterozygous for c.569G>A. Note that family 4 has two affected siblings, but parents were not carriers of the variant in blood DNA, suggesting parental gonadal mosaicism. In family 6 and 7, the affected individuals marked with + were heterozygous for the c.477+1G>A variant. Note that in family 6, gray color indicates mosaicism in blood DNA for the variant. In family 7, the older half-brother, mother, and maternal grandmother had suggestive clinical features but variant testing is pending. The pedigree of individual 3 is not included in this article as her family members did not agree for its publication. b Clinical pictures of proband 2-II:2, nine years. c Proband 4-II:4, 15 years. d Proband 1-III:4, three years, and his father 1-II:6 (e) and uncle 1-II:5 (f). g Half-siblings from family 7: aged 6 years and nine months and 2 years and 7 months, respectively. Note the variable severity of limb deformities. Written consent was obtained for publication of the photographs.
Fig. 3: Lateral radiographs and sagittal MRI of the lumbar spine.
a Radiograph of Patient 1-III:4 at age 3 years showing mild platyspondyly. b MRI of Patient 3 at age 7 years showing mild platyspondyly with irregular end plates and lumbosacral lordosis. c Radiograph of Patient 4-II:1 at 18 years of age showing mild platyspondyly with endplate modification. d Radiograph of Patient 4-II:4 at 15 years of age showing mild modification of vertebral end plates. Platyspondyly is not seen. e Radiograph of Patient 5-III:1 at age 7 years showing mild platyspondyly with severe lumbosacral lordosis and irregular end plates, and short neural arches of the lower lumbar spine and spinal canal stenosis. f Radiograph of Patient 6-II:1 at 7 years old, showing subtle irregularities of the vertebral end plates. g Radiograph of Patient 7-III:2 at 8 years showing vertebral bodies with central notches and anterior ossification defects. h Radiograph of Patient 7-III:3 at 4 years showing increased lumbosacral lordosis and vertebral bodies with anterior ossification defects and irregular end plates.
Fig. 4: Radiographs of the pelvis.
a Patient 1-III:4, radiograph at 3 years; showing unossified capital femoral epiphyses, and metaphyseal irregularities of the proximal femora with short femoral necks and coxa vara, and irregular acetabula. b Patient 2-II:2, radiograph at 9 years; showing flat, irregular capital femoral epiphyses, metaphyseal dysplasia of the proximal femora with short necks and coxa vara, and normal acetabula. c Patient 3, radiograph at 11 years; showing short femoral neck, severely flat capital femoral epiphyses, severe coxa vara and shallow acetabula, together with high-rising greater trochanters. d Patient 4-II:1, radiograph at 25 years; showing short femoral necks with mild coxa vara. The capital femoral epiphyses are not deformed, while the joint spaces are narrow. e Patient 4-II:4, radiograph at 15 years; showing severely flat capital femoral epiphyses and short femoral necks with mild coxa vara and high-rising greater trochanters. f Patient 5-III:1, radiograph at 10 years; showing short femoral neck, severely flat capital femoral epiphyses, severe coxa vara, and shallow acetabula. g Patient 5-II:2 the radiograph at 42 years showing flat capital femoral epiphyses, short femoral necks with mild coxa vara and high-rising greater trochanters, and narrow joint spaces. h Patient 6-II:1, radiograph at 7 years; showing short femoral necks, very small and flat capital femoral epiphyses, metaphyseal irregularities of the proximal femora, and severe coxa vara. i Patient 7-III:2, radiograph at 6 years 10 months; showing short femoral necks, flat capital femoral epiphysis of the left and delayed capital femoral epiphyseal ossification of the right, and severe metaphyseal irregularities of the proximal femora. j Patient 7-III:3, radiograph at 2 years and 3 months; showing short femoral necks, delayed ossification of both capital femoral epiphyses and severe metaphyseal irregularities of the proximal femora.
Fig. 5: Knee radiographs.
a Patient 1-III:4, radiograph at age 3 years; showing irregular metaphyses and small epiphyses of the distal femora and proximal tibiae with mild genu varum. b Patient 3, radiograph at age 11 years; showing genu varum and defective ossification of the medial aspect of the proximal tibial epiphysis. 8-plate guided growth was inserted to restore the knee deformity. c Patient 4-II:1 radiograph at age 18 years; showing mild epiphyseal dysplasia. d Patient 4-II:4, radiograph at age 15 years; showing mild epiphyseal dysplasia. e Patient 5-III:1, radiograph at age 3 years; showing small epiphyses and metaphyseal irregularities of the distal femora and proximal tibiae with bilateral genu varum. f Patient 5-II:2, radiograph at age 42 years; showing premature degenerative joint disease. g Patient 6-II:1, radiograph at 7 years; showing metaphyseal changes of the knees and mild genu varum. h Patient 7-III:2, radiograph at 7 years; showing mild metaphyseal changes and mildly flat epiphyses of the knees, genu valgum. 8-plate guided growth surgery partially restored severe genu valgum. i Patient 7-III:3, radiograph at 2 years; showing mild metaphyseal changes, mildly flat epiphyses of the knees and genu varum.
Family 1Family 1 had eight affected individuals (Fig. 2a, d–f). Proband I-III:4 born at full term with a normal perinatal history, presented with short stature and bilateral progressive genu varum at the age of 3 years (Fig. 2d). Radiographs showed unossified carpal bones, mild platyspondyly, coxa vara, and epimetaphyseal changes of the hips and lower extremities. The proband’s father (II:6) had severe genu varum at age 41 years (Fig. 2e), while the paternal uncle (II:5) had unilateral genu varum at 35 years (Fig. 2f). Individuals I:2, II:5, II:7, III:3 and III:6 all had genu varum, but further clinical data was not available. Individual I:3 was reportedly healthy without skeletal manifestations, and his height was 162 cm (−2.0 SDS). However, radiographic evaluation was not possible. All affected members in this family were heterozygous for a missense variant in RPL13, NM_000977.3: c.548G>A, p.(Arg183His).
Family 2Family 2 had one affected individual (2-II:2) (Fig. 2a, b). He was born to a non-consanguineous couple and had an unaffected twin sister. At 9 years old, he experienced walking difficulties, pain in his left leg, and general muscle weakness. Radiographs showed delayed carpal ossification, thoracic kyphosis, irregular vertebral endplates, mild platyspondyly, coxa vara, and epimetaphyseal changes of the hips and lower extremities. The patient had a de novo heterozygous missense variant in RPL13, NM_000977.3: c.548G>A, p.(Arg183His).
Family 3The proband in family 3 is a 20-year-old female, born to non-consanguineous parents after an uneventful pregnancy. She presented with bowed legs at an early age, clinically visible as genu varum from around five months of age. She underwent several surgeries on both lower extremities during childhood including 8-plate guided growth treatment (Fig. 5b). She had short stature, and radiographs showed small scaphoid and lunate, irregular vertebral endplates, mild platyspondyly, and severe coxa vara, as well as epimetaphyseal changes of the hips and lower extremities. She was heterozygous for RPL13 variant, NM_000977.3: c.569G>T, p.(Arg190Leu).
Family 4Family 4 had two affected siblings, born to healthy first cousins of Turkish origin. The proband (4-II:4, Fig. 2a, c), a 24-year-old male and his 31-year-old sister (4-II:1) were born after normal term pregnancies with reportedly normal birth length and weights. The proband’s clinical manifestations were short stature, pectus excavatum, pes planus, and mild scoliosis. He complained of painful ankles and knees since the age of four, which limited his mobility and participation in sports activities. Skeletal examination showed irregular vertebral endplates, mild epimetaphyseal dysplasia of the knees, as well as severely flat capital femoral epiphyses and short femoral necks with mild coxa vara and high-rising greater trochanters.
His sister (4-II:1) had reoccurring patellar dislocation in the left knee, which required patellar stabilization surgery at eight years. She experienced pain in the knees, legs, and lower back. Radiographs showed mild platyspondyly, irregular vertebral endplates, mild lumbosacral lordosis, short femoral necks with mild coxa vara and mild epiphyseal dysplasia of the knees. She has a healthy three-year-old daughter, delivered by cesarean section.
Both siblings were heterozygous for a missense variant, RPL13, NM_000977.3:c.569G>T, p.(Arg190Leu). This variant was absent in both parents’ blood DNA, suggesting gonadal and/or somatic mosaicism in one of the parents. Parenthood was confirmed by kinship estimates from WGS using single nucleotide polymorphism data. Given that joint dislocations have not been previously reported in SEMD-RPL13, we conducted an analysis of both single nucleotide and structural variants for all known skeletal dysplasia genes according to Genomics England PanelApp gene list. Our investigation did not reveal any other potential disease candidate variants.
Family 5The proband in family 5 (5-III:1) was a 24-year-old female born to non-consanguineous parents of Swedish/Finnish origin at full term. She had severe bilateral genu varum at birth and developed short stature postnatally. Radiographs showed delayed carpal bone ossification, mild platyspondyly with severe lumbosacral lordosis, coxa vara, and epimetaphyseal changes of the hips and lower extremities. She was treated with recombinant growth hormone (GH) from ages five to 15 years with a dosage of 0.06 mg/kg per day and IGF-1 levels around 400μg/L. Her initial basal serum IGF-1 level was 58 μg/L (normal range for age and sex 40–310 μg/L) and her initial height was −3.1 SDS. While her height improved during the first year of treatment, to −2.25 SDS, her final height was −3.1 SDS, indicating that GH treatment probably had no significant effect on the final height.
Her mother (5-II:2), a 64-year-old female of Swedish origin, had milder skeletal abnormalities than her daughter, including mild short stature (height 155.6 cm, −1.2 SDS) and genu varum that became less prominent with age. At 56 years, she underwent hip replacement surgery due to degenerative joint disease. Her radiographs showed mild platyspondyly with irregular endplates, flat capital femoral epiphyses and short femoral necks with mild coxa vara and high-rising greater trochanters, narrow joint spaces of the hip and degenerative joint disease in the knee. At the age of 63, she was diagnosed with rectal cancer. Both individuals were heterozygous for a missense variant, RPL13, NM_000977.3: c.548G>A, p.(Arg183His).
Family 6Patient 6-II:1 was born to healthy non-consanguineous parents of Hispanic origin. In the first year of life there was a noticeable growth deceleration, and during the initial evaluation at 1.5 years of age, radiographs revealed the presence of spondyloepimetaphyseal dysplasia. Both large and small joints were observed to have joint laxity. Subsequent radiographs at 6.5 years showed normal carpal bone ossification, mildly irregular vertebral endplates, flattening of the capital femoral epiphyses with coxa vara and epimetaphyseal changes of the lower extremities. The patient was heterozygous for a splice variant in RPL13, NM_000977.3: c.477+1G>A, while his asymptomatic father was found to carry the variant in 29% of his alleles in peripheral blood DNA, consistent with a somatic mosaicism.
Family 7Family 7 has two affected maternal half-siblings born at term after normal pregnancies. At age 6 and 2 years, respectively, they presented with progressive short stature and lower limb deformities (Fig. 2g). The older brother (III:2) had genu valgum, and the younger sister (III:3) had genu varum and coxa vara (Fig. 5h, i). Both siblings underwent 8-plate guided growth knee surgery. Their mother (II:1) has not been clinically evaluated, but she reportedly has a history of chronic mild hip joint pain. The maternal grandmother (I:2) has experienced hip joint pain since childhood. At age 35, she underwent bilateral hip replacement surgery, and her right hip has since undergone three revision surgeries. She also had surgical procedures on her right knee, however, there are no records available regarding the procedure that was performed. Although she is currently pain-free, she walks with a gait. An older maternal half-brother (III:1), aged 18, also reportedly has hip pain and walks with a limp. The siblings were heterozygous for a splice variant in RPL13, NM_000977.3: c.477+1G>A. Confirmatory molecular testing of the mother, grandmother, and maternal half-brother is currently ongoing.
Molecular findingsTwo variants in RPL13 (NM_000977.3) were identified, c.548G>A, p.(Arg183His), and c.569G>T, p.(Arg190Leu). These variants were detected in two unrelated families and segregated with the phenotype of SEMD-RPL13. None of the variants were present in gnomAD population database or in our in-house databases of 1455 exomes24 and 6781 exomes and genomes (Karolinska University Hospital Stockholm). One previously described variant, occurring at the same highly evolutionary conserved position 183, p.(Arg183Pro), has been reported in an individual with SEMD-RPL136. Based on the ACMG-AMP guidelines, we consider the two variants likely pathogenic. Three patients, including two half-siblings from family 7, were confirmed heterozygous for the previously reported splice variant c.477+1G>A, classified as pathogenic. The father in family 6 carries the variant in 29% of his alleles in peripheral blood DNA, making him a mosaic carrier for the genetic alteration. Functional studies by Le Caignec et al. demonstrated that this variant induces an aberrant mRNA, containing 54 bp of intron 5, resulting in the insertion of 18 amino acids within the protein6.
Predicted structural and functional effects of the eL13 variantsAll known eL13 missense mutations are found in H7, as shown in Fig. 1c, d. The residues in H7, except for Arg190, are highly evolutionary conserved with nearly maximal ConSurf-DB scores (8 out of 9)25 (Figure S1). H7 is amphipathic and has two distinct “faces”, one being primarily apolar/hydrophobic and interacts with 60S ribosomal proteins (eL27a and eL36), and the other polar/hydrophilic containing positively charged arginine residues (Arg183, Arg186, and Arg190), which bind to negatively charged phosphates (nucleotides G979, G980, and U981) in a 10-mer hairpin structure (AUGAAGGUGA) from ES9L 28S rRNA expansion segment. While interactions with eL27a and eL36 are primarily hydrophobic and involve H7 apolar residues (Ala178, Ala185), the mutated Arg183 and Arg190 are side by side, on the opposite surface of H7, and come in contact with phosphate groups, resembling “arginine forks”, previously described by Chavali et al.26. Notably, the recognized 28S rRNA segment forms a double-stranded RNA hairpin stabilized by a non-canonical G·U wobble pair configuration (U975-G980), which is known to mediate highly specific protein-RNA recognition27. This hairpin structure is further recognized by Arg190 binding to unpaired G979, and a distinct bend is present in helix 7, allowing interactions between the G·U wobble pairs and the protein, as previously described28. All currently known variants are in a 30 Å sphere where eL13 interacts with 28S rRNA through two specific RNA-binding sequences, as seen in Fig. 1 and Figure S1C. These amino acid substitutions can potentially disrupt the interaction of eL13 with 28S rRNA and the neighboring ribosomal protein through different mechanisms: (1) Arg183His introduces a shorter and bulkier side-chain with pH-variable charge; (2) Arg190Leu removes the positive charge of H7 that stabilizes RNA binding; (3) Arg183Pro introduces a kink changing the orientation of H7, and (4) Ala178Glu introduces negative charges that would repel the negatively charged RNA. In contrast to these missense mutations, the insertion of 18 amino acids after Asn159, previously reported by le Caignec et al, located approximately 30 Å away, targets the non-conserved extended loop that weakly binds ES7L 28S rRNA (nucleotide A509). This extended loop acts as a physical spacer between the N- and C-terminal RNA-binding regions of eL13. Therefore, this insertion would also disrupt the proteins’ interaction with rRNA within the missense cluster by pushing H7 further away.
Comments (0)