
Remember me
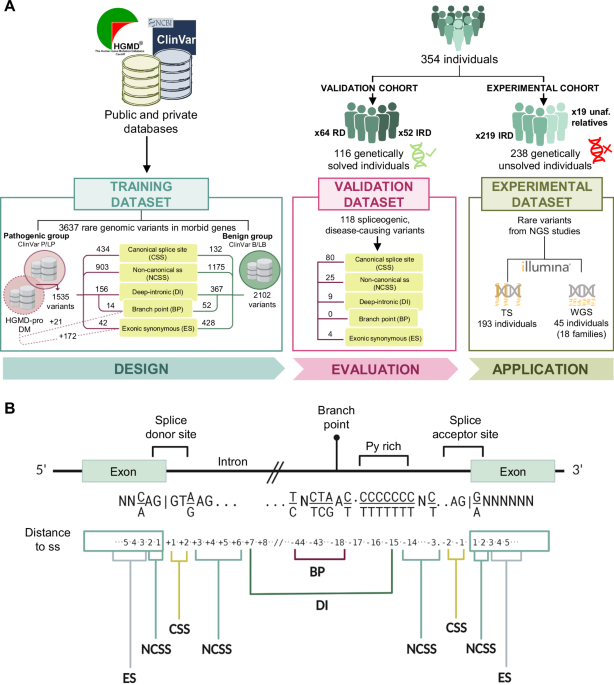
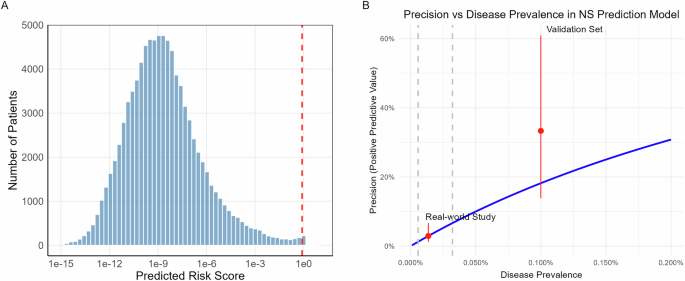


To investigate the characteristics of GCR in the Chinese population, we re-analysed variants included in ChinaMAP covering the deep WGS of 10,588 individuals14. After quality control, we extracted a total of 140,109,159 variants. To uncover the signature of GCR, 2904 phenotypes corresponding to 2464 AR genes exhibiting Mendelian inheritance were extracted from Online Mendelian Inheritance in Man (OMIM) as candidate genes, where genes associated with syndromes caused by large segmental duplications or chromosomal variations were discarded. The 12.7 million candidate gene overlapping variants were extracted, including 11,826,063 SNPs and 898,110 indels. These variants were subjected to a carefully designed workflow to classify variants as deleterious (Fig. 1a and Supplementary Fig. 1, see “Methods”). We then estimated GCR based on these deleterious variants. Meanwhile, the disease-associating variants were categorised into groups depending on whether treatments were available for the associated conditions/diseases.
Fig. 1: Distribution of variants and identification of AR disease-causing variants.
a A schematic flow of classifying variants extracted from ChinaMAP. Variants that passed the initial QC are subjected to downstream annotation depending on the nature of genomic loci (protein-coding, intron, splicing junctions, UTR, etc.). The annotated pathogenic variants in protein-coding regions are further divided into the following sub-categories: treatable, untreatable, either associated with LLPS or not, where the number of corresponding genes is shown. To compare the prediction accuracy of missense variants with Zhu et al.’s method, all P/LP and B/LB missense variants extracted from ClinVar (v20221113) are taken as true sets to generate AUC-ROC plot by the R package pROC taking the number of predict tools as threshold. b Comparisons of the count of passed-QC variants in distinct ethnic groups included in ChinaMAP and gnomAD databases. c The detailed comparison of variant numbers across chromosomes between gnomAD and ChinaMAP. d Shared SNPs of East Asian populations between ChinaMAP, gnomAD and WBBC. e Principal component analysis based on allele frequency of all studied populations.
We identified 19,484 deleterious variants associated with AR diseases. Among these, 3409 protein-coding variants were annotated by ClinVar database. Except for 19 manually curated variants, 14,775 and 1281 were predicted by our workflow as deleterious missense and deleterious nonsense, respectively (Fig. 1a). Compared to the previously estimated GCR19, the GCR reported in our study exhibited a moderate correlation, potentially caused by the intrinsic difference of variants selection using different strategies, cutoffs and prediction tools (Supplementary Figs. 2 and 3). After a systematic comparison, we confirmed that our strategy performed better in predicting P/LP variant as indicated by AUC- ROC (Fig. 1a), and that predicting tools and corresponding thresholds used in this study were strongly evidence-based and widely used, leading to a theoretically lower false discovery rate and a less biased variant set (Supplementary Table 1 and Supplementary Fig. 4). Indeed, GCRs reported here were more consistent with previous designs (Supplementary Fig. 5)20. Interestingly, 142 coding variants within 51 AR disease genes were annotated to be involved in LLPS. Among these variants, 65 (53.72%) were catalogued in ClinVar, 55 (45.45%) were deleterious missense variants, and only one was LoF. In contrast, 8.4 million non-coding variants, accounting for 92% of the non-coding variants, were enriched in the intronic regions of 1807 AR disease-causing genes. Unexpectedly, 49 genes were predicted to participate in LLPS. This suggested a promising yet not well-studied direction to disclose the affected regulatory processes beyond misfolded proteins.
As protein-coding sequences only comprised a small portion of the entire genome and many proteins are not targetable by small molecules, we retrieved non-coding variants associated with clear disease-causative genes to explore their potential functional characteristics. Surprisingly, 5.01% and 4.80% of these non-coding variants were enriched within the untranslated regions and up-/downstream of 1891 well-characterised disease-causative genes, respectively. Approximately 36,655 non-coding variants, accounting for 0.45% of the genes associated non-coding variants, were particularly enriched at splicing junctions. Around 45% of these variants were canonical splicing sites, and hundreds of sites were validated by the expression profiles in GTEx (102) (Supplementary Fig. 6) and supported by ICGC (802). However, only 1908, accounting for 5.2% variants, were predicted as splicing-altering sites by SpliceAI (438 acceptor gain, 315 acceptor loss, 269 donor gain, 886 donor loss)21. More interestingly, non-coding variants were more enriched in disease-causative genes than non-OMIM genes (Wilcoxon test, p < 2.2e-16). These observations implied that post-transcriptional regulation was deeply involved in the pathogenesis apart from that caused by altered protein products.
WBBC and gnomAD were used to assess the power of ChinaMAP data. Indeed, ChinaMAP detected more than 12 million single nucleotide polymorphisms (SNPs), which was over two-fold on average of that for each chromosome in the East Asian ethnic group (EAS) reported by gnomAD (Fig. 1b, c), thus enabling the generation of a much broader and more comprehensive view of the Chinese population. A further comparison of the mutation spectra between ChinaMAP, WBBC and EAS demonstrated that only 43% of gnomAD EAS and 42% of WBBC variants were shared with ChinaMAP, respectively, indicating potentially distinct genetic compositions among datasets (Fig. 1d). Further analysis of allele frequency confirmed that such minor discrepancy did not affect the separation of distant populations (Fig. 1e).
Estimation of gene carrier rate enhanced by deep WGS in East Asian populationChinaMAP covered many diverse minority groups in China, presenting the most comprehensive variations of the East Asian population to date. A substantial number of variants were not detected in the gnomAD EAS population due to technical limits or smaller sampling sizes (Figs. 1b and 2a). Thus, exploiting the dataset to obtain a systematic comparison of GCR was carried out to demonstrate the necessity of WGS to gain improved insight into reproduction counselling. Despite the slightly distinct GCR across populations, many genes of which GCR ≥ 1/500 in different ethnic groups were shared by at least two populations (Fig. 2b and Supplementary Fig. 7), suggesting an ancestral origin that potentially underwent selections22. However, the GCR of DMGDH, CD36 and GJB2, the variants on which were linked to diseases Dimethylglycine dehydrogenase deficiency, Platelet glycoprotein IV deficiency, and Deafness (autosomal recessive 1A), respectively, exhibited substantial variations across ethnic groups. For instance, variants on gene CD36 that could cause Platelet glycoprotein IV deficiency were ranked at the top in East Asian groups. In contrast, its observed GCR was drastically lower in several other groups, confirming that diversity caused by potential population-specific selections existed between some groups (Fig. 2c). However, the rank of many genes remained unchanged in other ethnic groups compared to ChinaMAP. In addition, numerous genes with highly ranked GCR were also observed at the leading ends within several other populations, confirming that AR disease genes could commonly affect many ethnic groups other than the non-negligible diversity (Fig. 2b and Supplementary Fig. 7b).
Fig. 2: Unique characteristics of GCR estimated from ChinaMAP.
a Distribution of GCR estimated from all investigated public datasets. Bottom: The distribution of GCR estimated from clear AR disease-causing variants; The green dot and red square represent the GCR that covers 99% and 95% of studied genes, respectively. Top: The corresponding sample size of each ethnic group in the studied databases. b Hierarchical clustering of genes with GCR ≥ 1/500 in ChinaMAP are selected to compare with corresponding GCRs in other studied populations. c Top 30 genes with drastic GCR changes, the order of which is determined based on the maximum pairwise distance of GCR between populations for a given gene, are selected to demonstrate ethnic-specific GCR features (solid: higher GCR in the East Asian group; dashed-line: lower GCR in the East Asian group). Additionally, the relative rank change of selected genes compared to ChinaMAP is indicated by line type. d The overlap of gene lists between ChinaMAP-derived AR disease-causing genes and genes associated with rare diseases officially listed in China.
By comparing GCRs estimated from these datasets, we found that the sequencing depth (30X for ChinaMAP, 18X for gnomAD v3) and sample size affected the estimation of GCR in several manners (Fig. 2a). The sample size dictated the detection power, while the under-detected variants caused by insufficient sequencing depth could be compensated by increasing the sampling size23. We speculated that the partial congruence with previously estimated GCR of EAS in gnomAD was due to the insufficient detection of variants and potentially false claim of ethnicity. To prove this, we performed PCA analysis on the ChinaMAP, EAS of gnomAD and WBBC at allele frequency level (Fig. 1e) and GCR correlation test between distinct ethnic groups (Supplementary Fig. 8 and Supplementary Table 11) to estimate the impact of low-sequencing coverage and insufficient recovery of rare variants. Indeed, approximately 3.6 million SNPs detected in both ChinaMAP and WBBC were missing in gnomAD, a much larger number compared to that between gnomAD and these two datasets, implying that a lower detection rate of rare variants in the EAS group of gnomAD cohort (Fig. 1d and Supplementary Fig. 9).
After intersecting the selected genes with a list of rare diseases compiled by the Chinese Center for Disease Control and Prevention, we found very few overlapping genes, suggesting that many of the rare diseases lacked appropriate genetic dissections. Alternatively, this potentially indicated the inflation of GCR due to either the carrier effect or heterogeneous genetic composition that could impact the estimation of GCR (Fig. 2c). ChinaMAP uniquely enabled the recovery of genes with higher GCR (Supplementary Fig. 10), confirming that EAS in gnomAD had a distinct genetic background compared to ChinaMAP. We also observed that the estimated GCR of treatable and untreatable diseases across several ethnic groups surprisingly differed significantly (Supplementary Fig. 11). This reinforced the necessity of introducing reproductive counselling for preventative purposes, allocating resources to find novel drug targets, and carrying out innovative research for rare diseases.
Discovery of potential druggable targets for rare diseasesAlthough responsible genetic variants of a few neonatal conditions were characterised, many conditions lacked approved treatments, including effective drugs and clinical therapies (Fig. 1a). Most rare diseases are not treatable, calling for unavoidable attention to explore potential druggable targets for these untreatable diseases besides implementing preventative measures.
In addition, the prevalence of certain diseases was primarily dictated by population-specific genetic compositions. Most affected genes varied across the population. For example, genes UGT1A1, SERPINB7 and ABCG5 were responsible for Crigler-Najjar syndrome, palmoplantar keratosis, phytosterolaemia, exhibited significantly lower GCR in non-Asian ethics compared to that of the Asian ethnic group, suggesting heterogeneous GCR at the population level needed to consider a population-centric priority for the variable genes when implementing preventative strategies or diagnostic strategies6,12,24. For investigation, both Asian groups seemed to be more susceptible to diseases Platelet glycoprotein IV deficiency, Lissencephaly 5, Deafness, autosomal recessive 111, Dyssegmental dysplasia, Silverman-Handmaker type/Schwartz-Jampel syndrome, type 1 and Citrullinemia, adult-onset type II / Citrullinemia, type II, neonatal-onset which were caused by mutations in CD36, LAMB1, MPZL2, HSPG2 and SLC25A13 supported by a slightly higher GCR compared to other ethnic groups12,15, restating the importance of strategic and peculiar drug development to target affected population (Supplementary Fig. 12).
Growing evidence indicated that LLPS participated in various regulatory processes by forming membrane-less organelles in cells, including transcriptional and translational dysregulation in pathogenesis16,17,25. As diseases caused by mutations within the protein-coding genes only account for a minor portion of the polymorphism, and many roles of non-coding variants were not examined thoroughly due to their poorly investigated functional roles, we expanded the search scope to identify non-coding variants associated with well-annotated pathogenic genes and potentially dysregulated condensate alterations during LLPS. The rationale was that intrinsically disordered regions pervasively existed in protein sequences and played essential roles in various biological processes through regulating non-membrane organelles. In total, 265,009 variants, accounting for 3.23% of the functionally uncharacterised variants, were involved in LLPS by querying against the list of predicted LLPS associating variants16, implying the direction of future drug design and exploration. Indeed, mutations in myosin VIIA and filaggrin could impair their ability to form condensates, leading to subsequent dysregulation of forming motor protein clusters in stereocilia25 and assembling keratohyalin granule in keratinocytes, respectively26. However, the GCR distribution was not significantly different across populations (Supplementary Fig. 13). A closer examination of the genes that could be impacted by dysregulated LLPS further revealed that at least 60% of diseases without available treatments were associated with the non-membrane organelle forming biomolecules predicted by previously reported approaches16 (Supplementary Fig. 14a). The functional enrichment of genes involved in LLPS indicated that genes were associated with extracellular matrix related functions. By performing GO enrichment of 14,487 non-coding variants adjacent to protein-coding genes (UTRs and splicing regions, see “Methods”), we also found that non-coding variants most likely affected genes (83 genes in the top 10 GO) that were involved in amino acid metabolic processes in addition to sensory perception, indicating a potential direction, such as secretome, to search for drug targets. Additionally, we observed that 265 genes fell into the non-treatable category (Supplementary Fig. 14b).
Rescale pan-ethnic preventative interventionFinally, we leveraged the power of the WGS-based mutation profile to attempt to exploit its potential in guiding the implementation of preventative measures. The cumulative GCR across distinct populations was surveyed, and it was clear that the number of highly ranked AR-diseases-causing genes were restrained within a certain range regardless of the population-specific genomic composition (Fig. 3a), suggesting that these genes were essential and were potentially selected though they have undergone diverse selection. Based on such observation, genes with a GCR threshold over 1/200 or 1/500 were examined to impute the panel design strategy considering the theoretical prevalence of a disease.
Fig. 3: Design of pan-ethnic screening panel.
a The cumulative carrier rate (CCR) of the top 2000 AR disease-causing genes is visualised. The thresholds of GCR used to select genes are indicated by grey dashed lines. b The proposed screening panel with three tiers, the number of included genes (LLPS associated) are chosen based on suggested cutoff 1/200 and 1/500 GCR, and the top 1000.
We found that only 20 AR disease-causing genes were exclusively detected by ChinaMAP with a threshold GCR > 1/200 (Supplementary Fig. 7), and all genes prioritised by ChinaMAP were unexpectedly shared with other ethnic groups when the threshold was lowered to GCR > 1/500, potentially because of the aggregated allele frequency of multiple ethnic groups. The comparison of GCRs derived from differential genes and their rankings (Supplementary Fig. 12 and Supplementary Fig. 10) revealed that such difference was likely due to the sampling or intrinsically distinct population structures in the studied populations. These genes showed significantly higher GCR than those in European (EUR) and African (AFR) populations.
Next, a strategy with a three-tier design was proposed to optimise the practical feasibility and rationale (Fig. 3b). Tier 1 and Tier 2 included 144 and 340 genes showing 1/200 and 1/500 GCR in the Chinese population, respectively. However, when the top 1000 genes of each population were extracted for comparison, over 83.26% of AR disease genes were shared by at least two populations (Supplementary Fig. 7c), restating the population heterogeneity could be circumvented once sequencing cost became a minor factor to be considered. The genes in Tier 2 have included genes to be screened during the carrier screening. Indeed, 55% to 80% of genes formed by previously reported panel designs were covered by the proposed panel at Tier 2 (Supplementary Fig. 15 and Supplementary Table 8). However, AR diseases caused by copy number variations, such as in HBA1/HBA2, DMD, SMN1, F8, F9, IDS, MTM1, GLA, IL2RG and OTC etc., were excluded in our current analysis. Such genes could be selectively supplemented to augment proposed panels based on prevalence in the population.
For genes included in Tier 3, although the theoretical prevalence of their associated AR diseases was much lower in the current Chinese cohort, they could be considered as a complementary list for couples to consider during reproductive counselling and expand the capacity carrier screening panel to a pan-ethnic panel once sequencing cost became neglectable and more reliable medical interpretation could be accessed through advanced or specialised artificial intelligence.
Comments (0)