
記住我
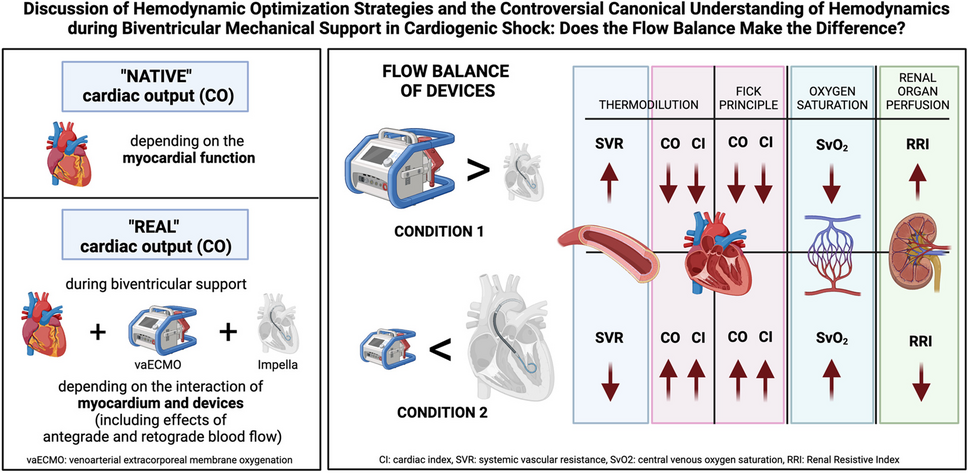


The GPS Registry (NCT05027685) is a multicenter, single-arm, observational study (Fig. 1). Up to 200 study centers from areas where the Paradise System (Recor Medical, Palo Alto, USA) is available will enroll patients, including but not limited to the European Union, the United Kingdom, Switzerland and Monaco. Additional locations may be added to the GPS Registry over time. Monitoring of study sites will be performed during the registry to assess continued adherence to the protocol and applicable regulations. The recommended follow-up is 5 years.
Fig. 1
Paradise ultrasound renal denervation system
All national and local approvals will be obtained prior to beginning the study. Any additional requirements imposed by the institutional review board/ethics committee (IRB/EC) or regulatory authority will be followed where appropriate. Annual IRB/EC renewals will be obtained throughout the duration of the registry as per local/country requirements. Written informed consent will be obtained from all patients or their legally designated representative, as defined by local regulations, in accordance with the Declaration of Helsinki.
The GPS Registry will be run under the guidance of a Steering Committee that will include international physicians with expertise in the areas of RDN, vascular medicine, hypertension, cardiology, interventional cardiology, radiology, and nephrology.
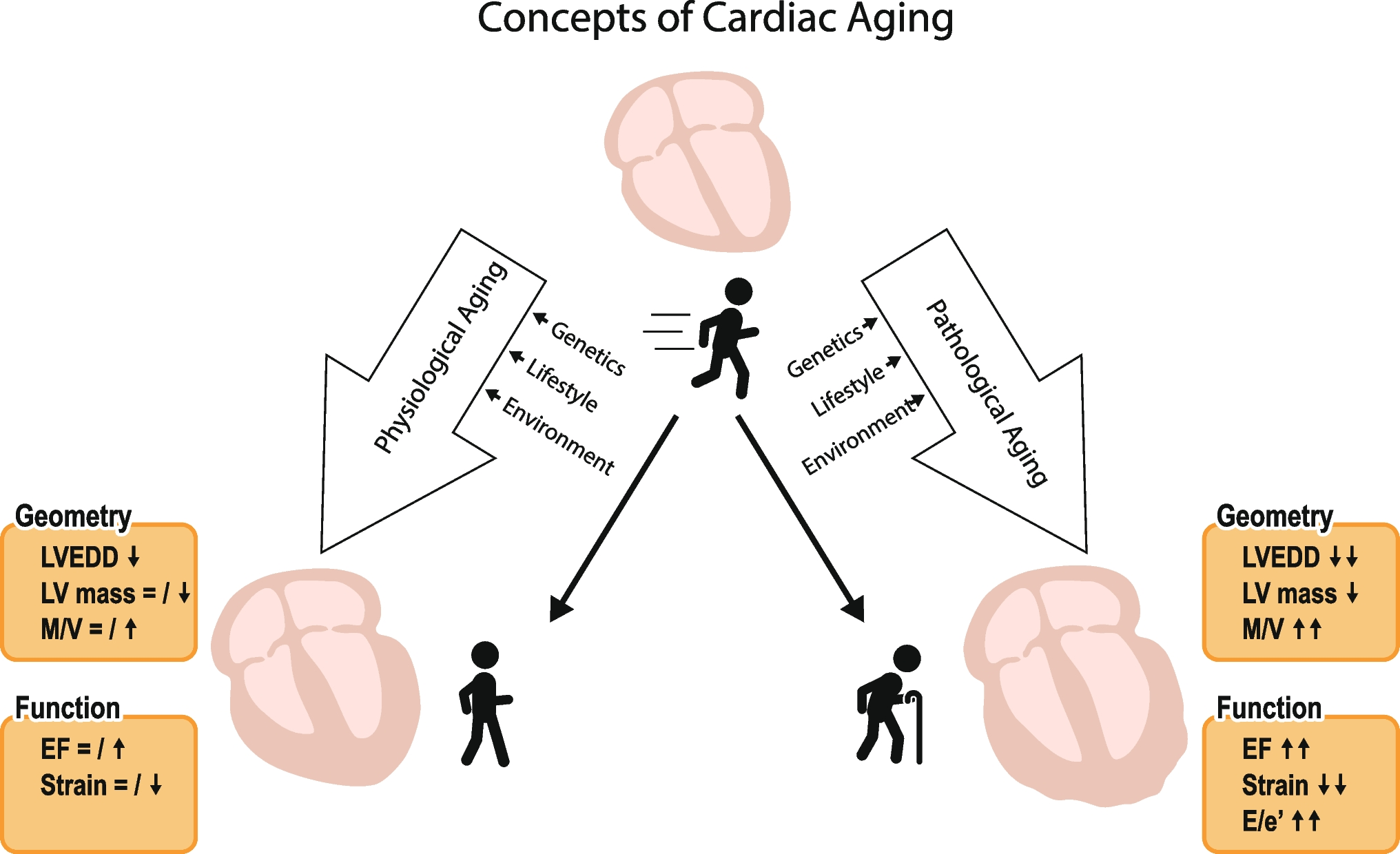
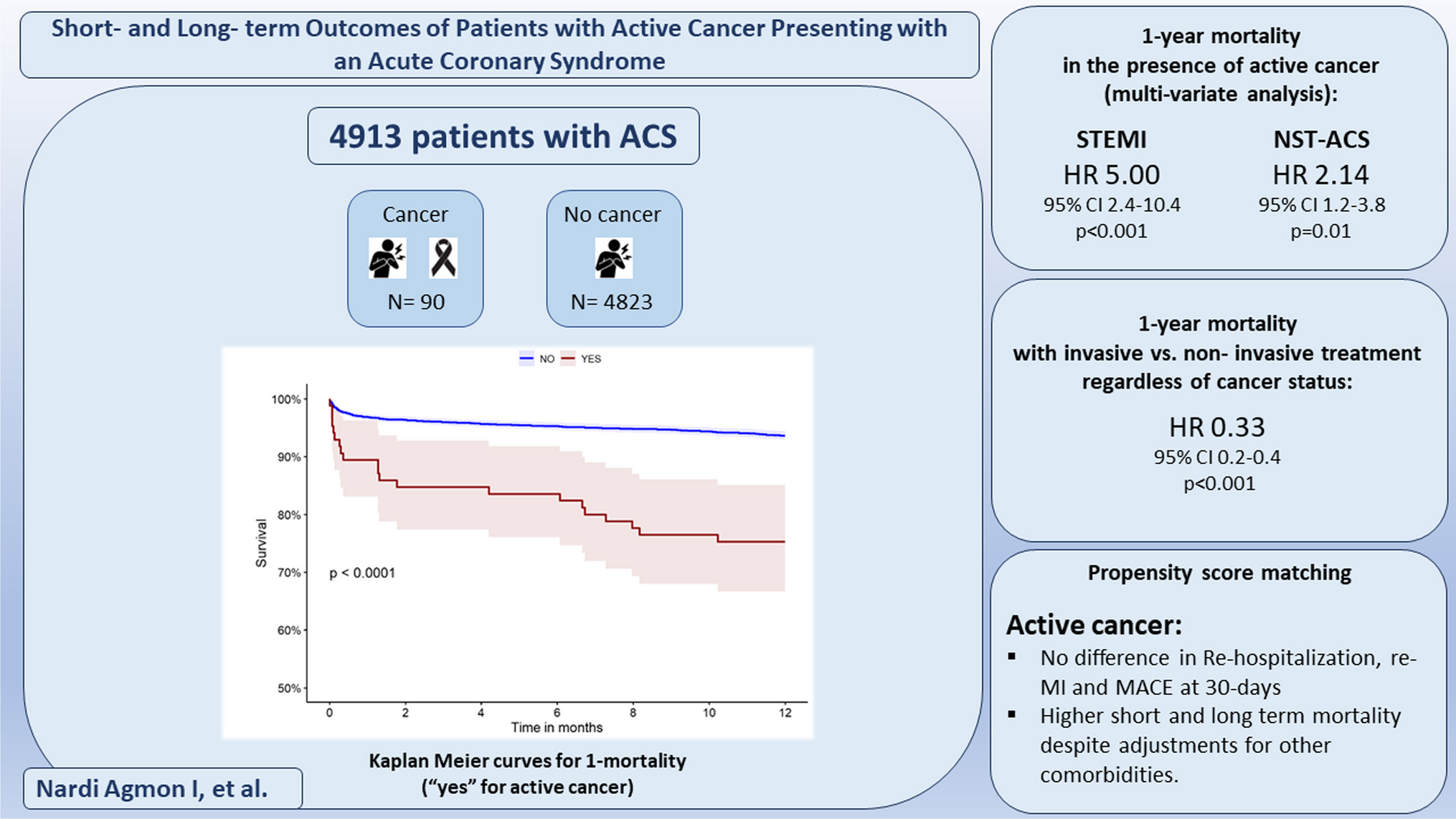
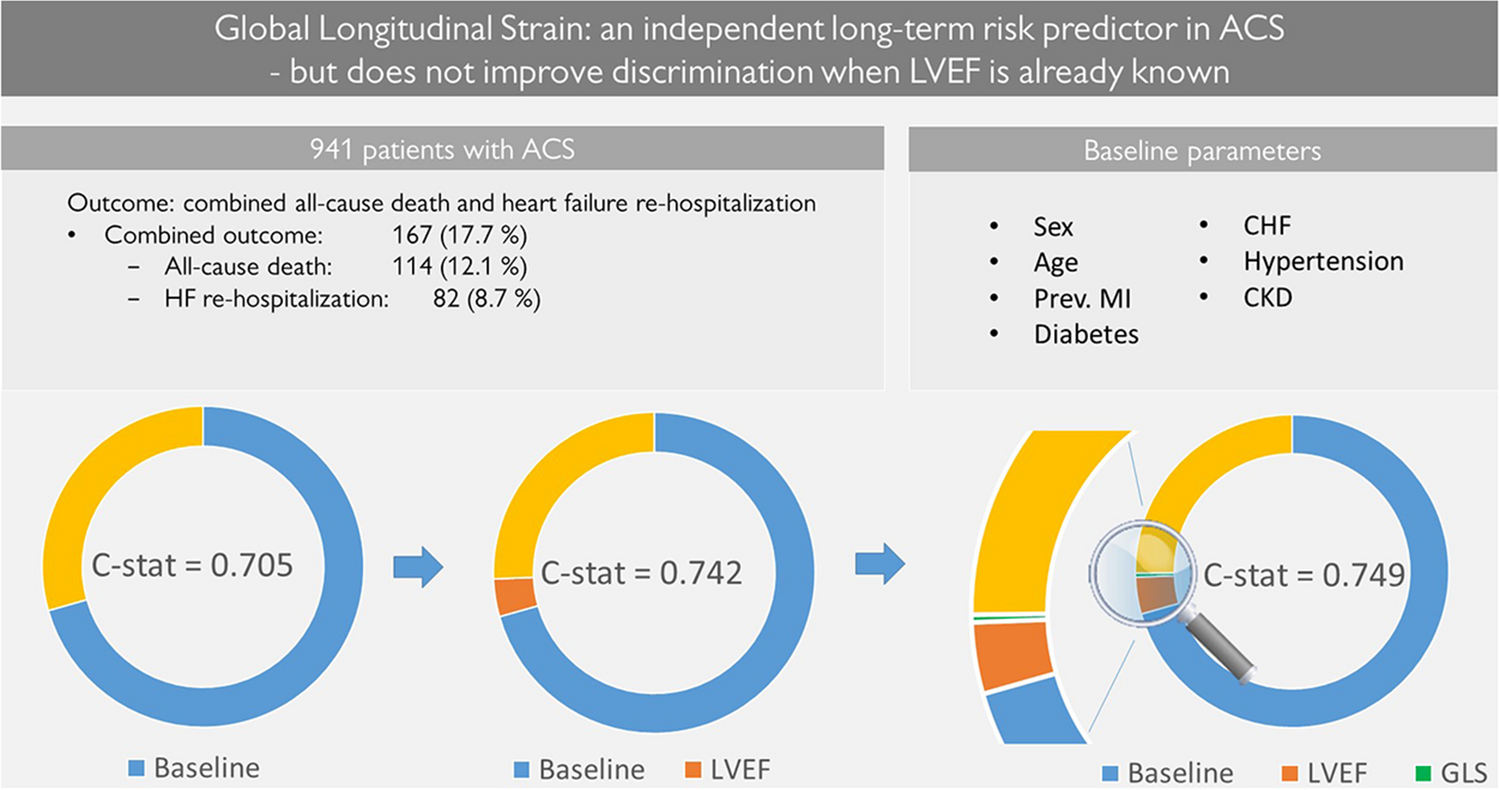
Study populationBased on the international guidelines that have been recently published, patients qualifying for renal denervation should have been unsuccessful in controlling their blood pressure with lifestyle changes and antihypertensive medications. Patients selected for uRDN with the Paradise System as per routine clinical practice on patient selection for renal denervation can be enrolled in the registry. The goal of the study is to collect real-world data on current clinical practices at each participating site. Full details of the study inclusion and exclusion criteria are shown in Table 1. For prospectively enrolled patients, the uRDN procedure is to be scheduled within 30 days of registry enrollment, while retrospectively enrolled patients need to have undergone uRDN within the previous 6 months.
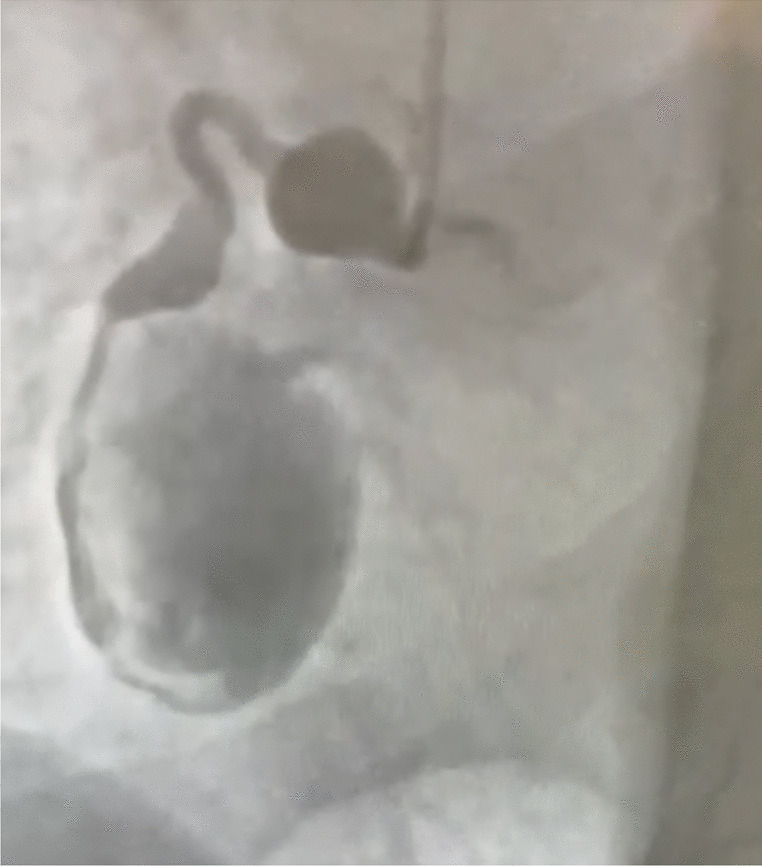
Table 1 Inclusion and exclusion criteriaRenal denervation procedureThe Recor Paradise System is a catheter-based system that delivers ultrasound energy to thermally ablate and disrupt the renal efferent and afferent sympathetic nerves, with the goal of achieving a reduction in systemic arterial BP and mitigating end-organ effects due to sympathetic over-activity. The system includes a single-use 6F catheter and an automated, portable, customized generator. The Paradise Catheter is intended to be employed in a catheterization laboratory under fluoroscopic guidance via femoral access only. The catheter consists of a through-lumen shaft with a cylindrical piezoelectric ceramic transducer located at the distal end of the catheter. The catheter has a distal balloon, which is pressurized by the generator to 1.5–2.0 atm using sterile circulating water.
The device's ultrasound transducer converts electrical energy into acoustic energy, which is then delivered radially through the cooling balloon into the renal artery. The pressurized balloon centers the ultrasound transducer within the artery, and the circulation of fluid serves as coolant to protect the endothelial and medial layers of the renal arterial wall. Each catheter has an embedded chip that communicates directly with the generator and specifies the power settings to be applied.
The uRDN procedure will be performed according to routine practice at each study site and consistent with the device’s instructions for use. As treatment is bilateral, specific angiographic requirements need to be met to deem anatomic eligibility. Measurements of the distal, mid, and proximal renal artery diameters are used to select the appropriate Paradise catheter balloon size. The treatment strategy requires operators to deliver a minimum of two (to three) sonications in each main renal artery and the first sonication should be delivered at a distance of at least 5 mm from the artery bifurcation. The Instructions for Use includes details for balloon sizing and treatment strategy regarding the number of emissions per artery and also includes instructions for additional sonications in case there are proximal bifurcations or accessory arteries. Physicians new to the technology will receive proctoring support for initial cases as needed.
Procedural support by a sponsor representative is available to all sites regardless of experience with the Paradise Renal Denervation System.
Assessments and data collectionFollow-up visits are scheduled at 3, 6, and 12 months after the uRDN procedure and every 12 months thereafter. Each visit will include medical review, measurement of office BP, medication name and dose). Ambulatory BP data (mean values, heart rate, standard deviation) obtained during routine clinical practice at study centers (including daytime, nighttime and 24-h BP) will be recorded and assigned to the nearest follow-up visit.
Other data collected during routine clinical practice will also be reviewed as part of patient follow-up, including any of the following: physical parameters including body weight, HR, seated and standing office BP; smoking status during follow-up, socioeconomic, and healthcare resource use parameters; electrocardiogram; non-invasive cardiac imaging; urinalysis including micro/macroalbuminuria; blood chemistry (serum creatinine, electrolytes, blood glucose and HbA1c, blood lipids); non-invasive renal artery imaging; any other cardiovascular procedures and treatments including glucose- and cholesterol-lowering drugs and antiplatelet/anticoagulant use, and the occurrence of any cardiovascular/cerebrovascular event (MI, HF, AF, etc.) and cardiovascular or non-cardiovascular death.
Home blood pressureAll clinical sites will be provided with validated, commercially available, validated telemetric home BP monitoring (HBPM) devices BPM Connect Pro, WITHING, Issy-les-Moulineaux, France) [19]. The collection of home blood pressure with this WITHING device is specific to the execution of the study and is not currently part of the sites' routine clinical practice. Prospectively enrolled subjects who elect to be part of HBPM data collection will be provided with a device and will be instructed to record their home BP according to the American Heart Association and American Medical Assocation [20] for seven consecutive days prior to the uRDN procedure. Over this period, subjects will take two measurements in the morning and two measurements in the evening. Consistent with current recommendations [20, 21], patients will be instructed to avoid smoking, caffeinated beverages, or exercise in the 30 min prior to home BP measurement, to take readings in a quiet room after sitting quietly for 5 min, to sit with their back straight and supported and feet flat on the floor with the arm supported on a flat surface and the upper arm at heart level. The same arm with the highest BP determined previously will be used for all home BP measurements, and at least two measurements should be taken 1–2 min apart before taking medications in the morning, and before taking medications and before dinner in the evening. All home BP measurements will be transmitted automatically via cellular or wifi signal by the HBPM device to a dedicated secured cloud-based platform (Study Pal; DELVE; www.delvehealth.com), which is GDPR 2016/679 compliant. The use of this integrated Global System for Mobile communication functionality removes the need and burden for participants to record home BP measurements in a diary.
Subjects will be reminded by automatic text message to measure their home BP if their home BP assessment is incomplete. Ideally seven- but a minimum of 3-day HBPM data (at least twelve valid measurements) are recommended prior to the procedure. Patients participating in home BP data collection will measure their home BP every 3 months during follow-up, ideally over 7 days of measurements with four readings per day prior each visit at the study site. If insufficient baseline home BP data are collected, no additional home BP data will be collected for that patient. Subjects who are unwilling and/or unable to comply with HBPM requirements will not take part in HBPM data collection but will continue to be part of the registry. Retrospectively enrolled subjects will not be provided with a HBPM device, and any available BP data (office, HBP, ABPM) from prior to the uRDN procedure will be collected retrospectively.
Patient-reported outcomesThe validated Short Form (SF)-12 questionnaire is a 12-item questionnaire that gathers data on patient functional health and well-being. The patient may elect to complete the SF-12 quality-of-life questionnaire as part of the study specific assessments. If patients agree to complete the questionnaire, they will be asked to complete the questionnaire in their local language at enrollment, at 3, 6, and 12 months after the uRDN procedure, and annually thereafter.
SafetyAll centers participating in the GPS Registry will use the Paradise uRDN system per their normal practice. Therefore, any device-related adverse events will be reported to the appropriate regulatory authorities through the Recor Medical vigilance reporting process. Underlying diseases/pre-existing conditions will not be reported as adverse events unless there has been a substantial increase in the severity or frequency of the problem that cannot be attributed to natural history during the course of the registry. The assessment of safety includes but is not limited to collecting the following events: incidence of new onset end-stage renal disease (eGFR < 15 mL/min/m2 or need for renal replacement therapy), significant decline in renal function, new renal artery stenosis > 70% by CTA/MRA, incidence of renal artery perforation or dissection requiring intervention, incidence of hospitalizations for hypertensive crisis or symptomatic hypotension.
Data collection and storageSubject data will be collected into an electronic data capture system. The data will be monitored and audited for data collection to assess continued compliance with the protocol and applicable regulations to ensure the integrity of the data. Data processing will be performed in compliance with the European Union General Data Protection Regulation (GDPR) 2016/679, and all applicable national laws to ensure data governance policies and guidelines, including data ownership, data access, data sharing, and data use agreements are managed properly.
Sample sizeThe sample size for the Registry is based on the intent to characterize the safety and effectiveness data in a real-world patient population. The target is to enroll up to 3000 patients at up to 200 sites globally and follow them up to 5 years after enrollment. This sample size should provide a high degree of statistical precision on home and office blood pressure changes and a high probability of observing rare safety events; with a sample size of 3000 subjects, there is a 95% probability of observing events that occur at a population rate as low as 0.1%.
Statistical analysisEvery effort will be made to minimize the amount of missing data. Statistical analyses will be descriptive. Continuous variables will be summarized as mean ± standard deviations and median (range) with interquartile range. Categorical variables will be summarized as frequencies and percentages. Analysis of clinical effectiveness and safety (Table 2) data will be performed on the total study population and in clinically relevant participant subgroups (e.g., based on age, history of chronic kidney disease, heart failure, atrial fibrillation, sleep apnea, diabetes mellitus, etc.). An additional analysis will determine whether BP parameters can help predict patients that may be more likely to respond to uRDN. Subgroup analyses and multivariable analyses will be performed.
Table 2 Standard of care parameters collected, effectiveness and safety assessments
留言 (0)