
Remember me

Penile carcinogenesis follows 2 main etiologic pathways with respect to the role of human papillomavirus (HPV). About 50% of invasive penile squamous cell carcinoma (SCC) worldwide arises through the oncogenic actions of HPV, the other half arises independent of HPV.1,2 With the advent of HPV genotyping and the introduction of p16ink4a testing as a surrogate marker for transforming HPV infections in the past decades, research concentrated mainly on HPV-induced invasive SCC3–6 and precursor lesions7–9 from high incidence areas for penile SCC. In contrast, the heterogenous group of HPV-independent penile SCC received little attention.10 HPV-independent penile SCC arise often in chronic lichenoid dermatoses2,10–15 and features mutations in tumor suppressor genes TP53 and CDKN2A,2,16–19 characteristics shared by vulvar SCC. Vulvar carcinogenesis has many parallels to penile carcinoma, but a higher and more robust level of evidence for the diagnosis of precancers.20 In analogy to vulva,21 the most recent WHO classification urinary and male genital tumors calls HPV-independent squamous precursors differentiated penile intraepithelial lesions (d-PeIN), but contrary to vulva, they were not further subtyped.22 Small cohort studies7,9,23 did not yield enough data for a systemic classification of HPV-independent penile precursors. For vulva and cervix, at least 2 types of highly differentiated HPV-independent precursor lesions and pathways of carcinogenesis are recognized.24–26 Basal cell proliferations with near-normal cornification and TP53 mutations, and verrucous/verruciform p53 wild-type proliferations with variable hyperkeratosis and parakeratosis stand opposite undifferentiated HPV-independent precursor lesions in vulva, cervix27,28 and penis.29 In the past decades, during preparation of various manuscripts on penile cancer14–16,19,30,31 and penile lichen planus32 we noticed similar histologic variants of penile precursors. The aim for this paper was to establish a classification of HPV-negative penile squamous precursors based on histologic, immunohistochemical, and genetic characteristics. We initially planned a comparative analysis of 70 peritumoral HPV-negative penile squamous precursor and the matching invasive SCC but the close spatial association of precursor lesion and invasive SCC made it impossible to obtain a pure sample of peritumoral precursor lesions thus precluding a bona fide mutational analysis. We therefore correlated the histology of the precursor lesions and their p53 and 16ink4a expression pattern with the known mutational profile of the invasive SCC.19 In this paper, we followed evidence from precancerous lesions at other anogenital sites and proposed a simple reproducible subtyping/classification of HPV-negative penile precancers with a terminology common to vulvar and cervical carcinogenesis.
MATERIALS AND METHODSWe analyzed 88 HPV-independent invasive penile SCC from the archive of the Diagnostic and Research Institute of Pathology, Medical University Graz, Graz, Austria for which HPV genotyping and mutational analysis were available.16,19
HPV genotyping was performed as previously described with CHIPRON (CHIPRON GmbH), and based on the principle of reverse hybridization and the SPF10 primer set which amplifies a 65 bp region in the L1 open reading frame of the HPV genome. Multiplex polymerase chain reaction and duplex hybridization allows detection of DNA of 32 HPV subtypes including HPV high-risk subtypes 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59; probably carcinogenic HPV68; potentially carcinogenic HPV subtypes 26, 30, 34, 53, 66, 67, 69, 70, 73, 82, 85, 97; HPV low-risk subtypes 6, 11. Alternatively, HPV status was evaluated according to manufacturer instructions with Vision Array HPV Chip 1 (ZytoVision GmbH) covering 41 HPV genotypes (with additional HPV low-risk genotypes). The 50 cancer-relevant genes analyzed in the invasive SCC included ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAQ, GNAS, HNF1A, HRAS, IDH1, IDH2, JAK2, JAK3, KDR, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, and VHL.16,19
Not all archival cancer specimens contained peritumoral tissue, which led to exclusion of 10 SCC. Another 8 SCC showed very short segments of peritumoral squamous proliferation/hyperplasia of <3 rete ridges without atypia, mitotic activity, or abnormal maturation that were not classifiable as dysplastic. The peritumoral precursors of the remaining 70 SCC were evaluated for histology, and immunohistochemically for p16ink4a (Roche MTM Laboratories) and p53 (antibody clone DO-7; Dako) expression. In analogy to vulva25,33,34 and cervix,26 highly differentiated penile HPV-independent precancers were divided histologically into those with elongated rete ridges and atypical basal cells with near-normal superficial differentiation and those with cornified verrucous hyperkeratotic and parakeratotic and noncornified verruciform/papillary intraepithelial proliferations. p53 staining was divided into overexpression, defined as uniform nuclear staining of basal and suprabasal cells, a wild-type pattern when only individual basal keratinocyte nuclei stained, and negative/null-type staining. p16ink4a overexpression was defined as homogeneous continuous cytoplasmic staining of all dysplastic cells, a so-called “block” staining. Discontinuous, heterogeneous, and patchy (background) staining, independent of the percentage of positivity, was interpreted as negative for overexpression. The presence of lichen sclerosus and lichen planus was recorded. Characteristics of the precursor lesions were compared with histology and mutational profile of the adjacent invasive SCC.
Statistical analysis was performed using open source Jeffries amazing statistics Program JASP, version 0.16.4 (www.jasp-stat.org). Comparison between groups were made by χ2 test.
Institutional review board approval was obtained on November 27, 2018 (31-049 ex 18/19).
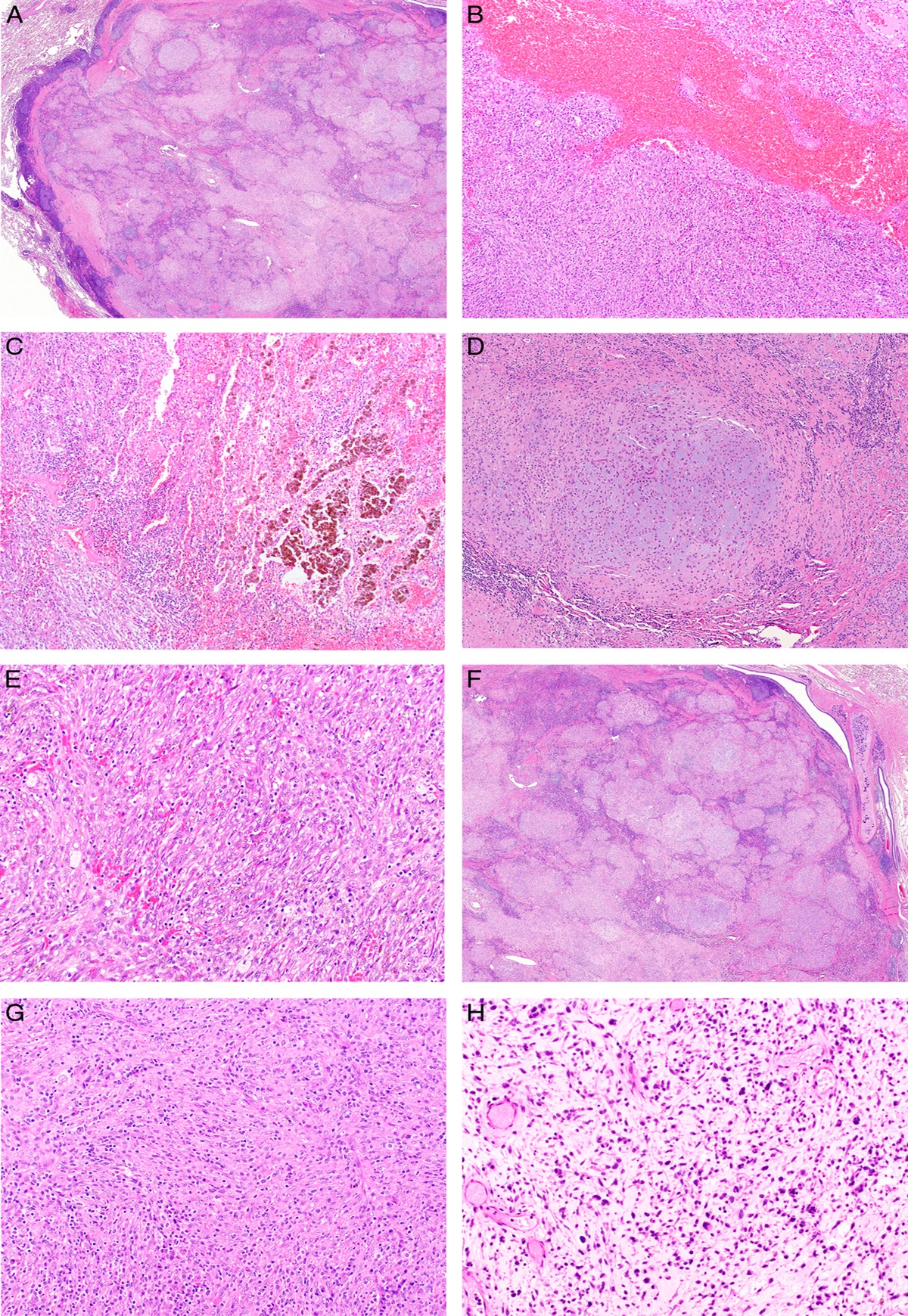
RESULTS Histology of Penile HPV-independent Precursor LesionsThe histologic patterns of precursor lesions in archival specimens of 70 HPV-independent penile SCC were analyzed. The most common pattern (n=42/70; 60%) featured a variously acanthotic epithelial proliferation with variously elongated rete ridges, but mostly normal maturation and superficial cornification. The basal and suprabasal keratinocyte layers showed an increased mitotic rate and significant keratinocyte atypia, with multifocal areas of premature squamatisation and occasional spongiosis (Figs. 1A, C, E). In all, 26/42 d-PeIN showed nuclear p53 overexpression (P<0.001; Figs. 1B, D, F) and 5/42 were p16ink4a positive. Focal mild pararkeratosis and hyperkeratosis in addition to a granular cell layer was present in d-PeIN, but stacks of parakeratosis and compact hyperkeratosis were absent. These lesions are referred to as d-PeIN throughout this paper. d-PeIN were identified adjacent to keratinizing G1-G2 (n=36; P<001), verrucous (n=1), clear cell (n=1), papillary/verruciform (n=3), and sarcomatoid (n=1) invasive SCC. Chronic inflammatory dermatoses were identified in 32/42 d-PeIN (28 lichen planus and 4 lichen sclerosus; P<0.001). For 6/42 d-PeIN no inflammatory background was identified. In 4 cases there was not sufficient tissue around the precursor lesion for evaluation of presence or absence of lichenoid dermatoses. In all, 10/42 SCC with d-PeIN showed either lymphovascular space (LVS) invasion or lymph node (LN) metastases (for summary, see Tables 1, 2, Fig. 2).
 FIGURE 1:
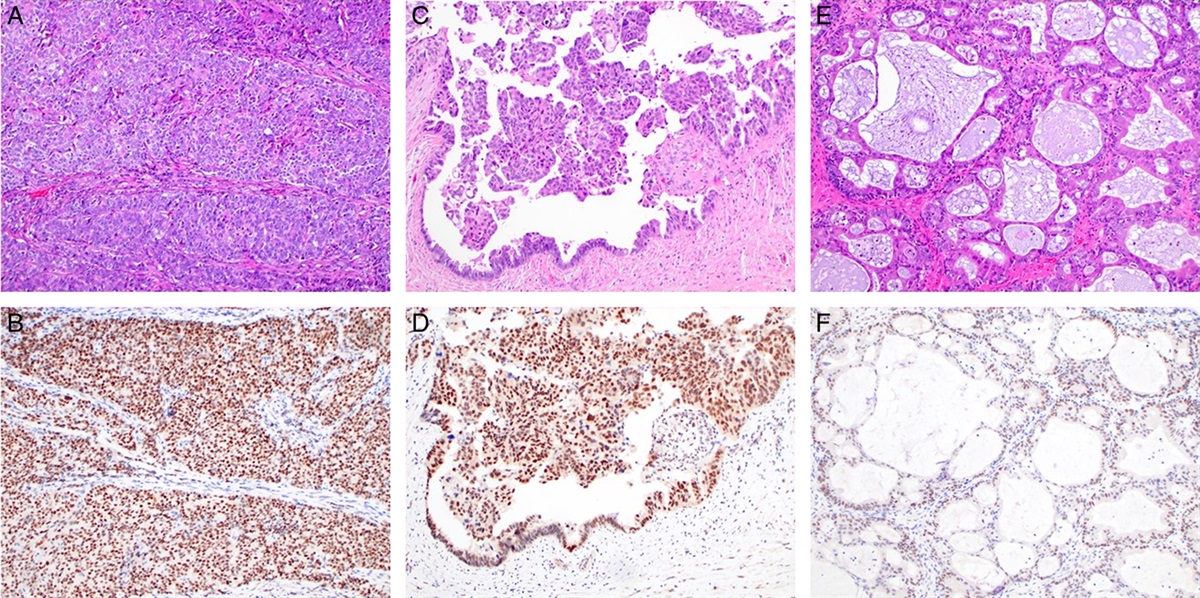
FIGURE 1: Differentiated HPV-independent penile precursors (d-PeIN). A, Hematoxylin and eosin–stained section of a d-PeIN adjacent to a keratinizing SCC with TP53 missense mutation and a truncating mutation in CDKN2A revealing elongated and branching rete ridges with premature squamatiziation, mitoses, and mild spongiosis. The dense lichenoid lymphocytic infiltrate is owed to the background of lichen planus. B, Immunohistochemical stain with antibody to p53 reveals focal nuclear overexpression in basal keratinocytes. C, Hematoxylin and eosin–stained section of a d-PeIN with elongated rete and normal cornification adjacent to a keratinizing SCC arising in gylcogenated mucosa without TP53 hotspot mutation but mutations in HRAS and SMARCB1. D, With prominent nuclear overexpression of p53 in basal and suprabasal keratinocytes. E, Hematoxylin and eosin stain of a precursor lesion adjacent to a SCC with a TP53 missense mutation and a trancating mutation in the CDKN2A gene. Note the focally atypical basal cell proliferation with mildly elongated rete ridges, individual mitosis, loss of glycogen but normal maturation and cornification. The basement membrane is focally thickened around pointed rete a dense lymphohistiocytic infiltrate. F, Mildly atypical basal and suprabasal keratinocytes show nuclear p53 over expression.
TABLE 1 - Summary of Statistical Analysis n (%) Type of precursor lesion d-PeIN, N=42 (100%) Verrucous/verruciform, N=21 (100%) Basaloid/undifferentiated, N=7 (100%) Significance (P) p53 overexpression 26 (63) 3 (14) 2 (29) <0.001 p16ink4a overexpression 5 (12) 1 (5) 1 (14) NS Chronic dermatoses (LP/LS) 32 (76) 7 (33) — <0.001 Adjacent invasive SCC Histology Keratinizing (G1-G2) 36 (86) 4 (19) 1 (14) <0.001 Verruciform/papillary in glycogenated mucosa 3 (8) 7 (33) 2 (29) 0.024 Verrucous in cornified skin/mucosa 1 (2) 10 (48) — <0.001 Combined verrucous or verruciform 4 (10) 17 (81) 2 (29) <0.001 Basaloid (G3) — — 3 (43) <0.001 Clear cell 1 (2) — 1 (14) NS Sarcomatoid 1 (2) — — NS LVS invasion 4 (10) 3 (14) 1 (14) NS LN metastasis 6 (14) — — NS Somatic gene mutations in SCC TP53 mutation 32 (76) 3 (14) 2 (29) <0.001 CDKN2A mutation 26 (62) 5 (24) 1 (14) 0.004 Concomitant TP53/CDKN2A 21 (50) 2 (10) 1 (14) 0.003 HRAS 5 (12) 7 (33) 1 (14) NS PIK3CA 5 (12) 7 (33) 1 (14) NS HRAS or PIK3CA 7 (24) 12 (57) 2 (29) 0.004LS/LP indicates lichen sclerosus/lichen planus; NS, nonsignificant.
 FIGURE 2:
FIGURE 2: Oncoplot demonstrating characteristics of precancerous lesions including histology, p53 and p16ink4a overexpression, presence of lichen sclerosus and lichen planus in comparison with p53 and p16ink4a overexpression and recurrently mutated genes of the adjacent invasive SCC. NA indicates not applicable.
The second type of precursor lesion (n=21/70; 30%) was characterized by a variously acanthotic verrucous squamous proliferation with variable amounts of compact hyperkeratosis, multilayered parakeratosis and absence of a granular cell layer. The plump epithelial rete showed minimal nuclear atypia and mitotic activity in basal cells. Overall, 14/21 precursor lesions were associated with invasive SCC that arose in the cornified mucosa of foreskin and glans penis and referred to as verrucous penile intraepithelial lesion throughout this paper (Figs. 3A–G). Another 7/21 precursors were noncornified, verruciform/papillary intraepithelial precursor lesions adjacent to SCC that arose in glycogenated squamous mucosa near the urethra and in the sulcus coronarius (Figs. 4A, B). The majority of 17/21 (65%) verrucous/verruciform precancers were identified adjacent to verrucous invasive SCC (P<0.001), only 4/21 occurred adjacent to keratinizing G1-G2 SCC. All verrucous/verruciform precursors were p16ink4a negative with one exception; only 3/21 showed nuclear p53 overexpression. In all, 3/21 SCC with verrucous/verruciform precursors showed LVS invasion (for summary, see Tables 1, 2; Fig. 2).
 FIGURE 3:
FIGURE 3: A–C, Verrucous acanthotic HPV-independent penile precursors without TP53 hotspot mutations. A, Hematoxylin and eosin–stained section of a massively acanthotic verrucous squamous precursor lesion without TP53 hotspot mutations with absent granular cell layer but thick stacks of parakeratosis and compact hyperkeratosis adjacent to a verrucous SCC with mutations in Notch1, HRAS, and SMAD4. B, The rete ridges are plump and irregularly elongated. C, The plump epithelial rete show minimal atypia, but increased mitotic activity in basal keratinocytes. D–G, Verrucous precancer adjacent to a verrucous SCC with 2 different TP53 missense mutations at low mutational frequency and a truncating mutation in the CDKN2A gene. D, Hematoxylin and eosin stain of verrucous squamous lesion with irregular rete, stacks of para and hyperkeratosis. E, Note the focal circumscribed basement membrane thickening and beginning subepithelial sclerosis with nuclear p53 overexpression of basal keratinocytes. Hematoxylin and eosin stain of another area of verrucous precursor lesion with irregular and elongated rete (F) with wild-type p53 staining of basal keratinocytes (G).
 FIGURE 4:
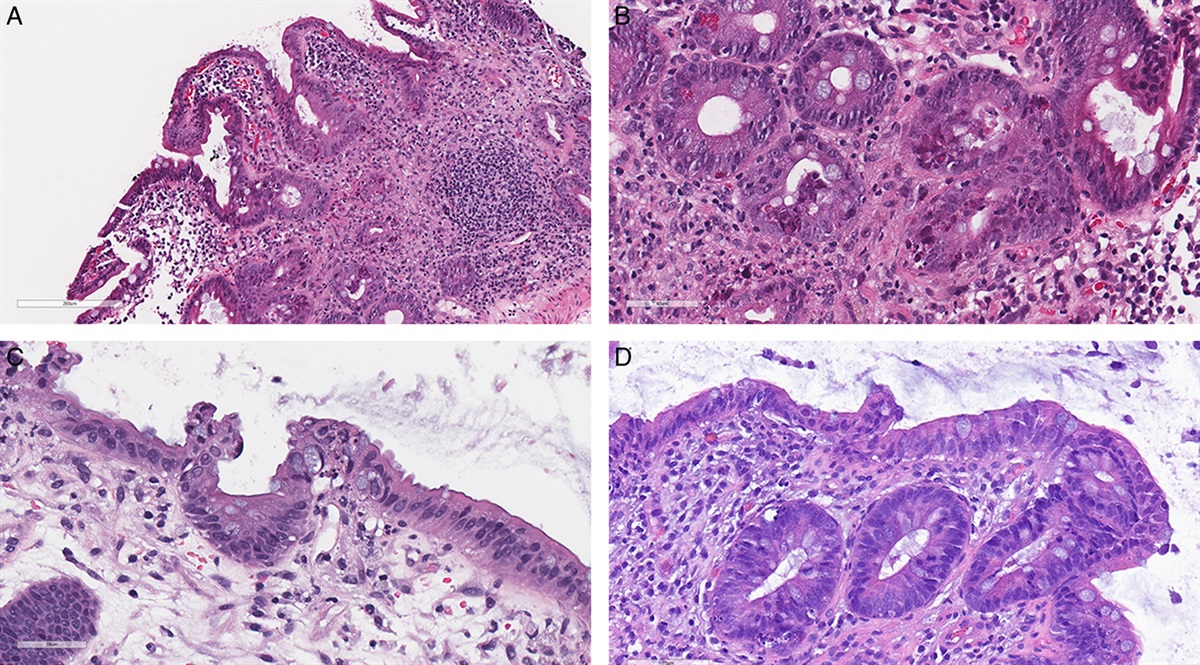
FIGURE 4: Noncornified verruciform and undifferentiated HPV-independent penile precursors. A, Hematoxylin and eosin–stained section of a verruciform noncornified peritumoral lesion of a papillary SCC without TP53 hotspot mutations but an FGFR3 mutation. B, Hematoxylin and eosin stain of another verruciform precursor lesion adjacent to a papillary SCC featuring 2 TP53 missense mutations and a truncating mutation in CDKN2A. Invasive SCC with clear cell differentiation (C) and hotspot mutations in TP53 and CDKN2A had an undifferentiated noncornified peritumoral precursor lesion (D). corresponding nuclear p53 overxpression (E) and weak cytoplasmic p16ink4a staining (F). G, Hematoxylin and eosin stain of an undifferentiated/basaloid precursor lesion with beginning cornification adjacent to a keratinizing SCC featuring 2 TP53 missense mutations and a PIK3CA mutation. H, but wild-type p53 staining in the peritumoral precursor lesion.
The rarest form of peritumoral precursor lesion was a full-thickness proliferation of undifferentiated, p16ink4a-negative cells in 7/70 (10%) (Figs. 4C–H). They were associated with keratinizing (n=1), papillary (n=2), basaloid (n=3), and clear cell (n=1) invasive SCC. Two of the undifferentiated basaloid HPV-negative precursor lesions showed nuclear p53 overexpression (Figs. 4C–F) and 1 of these also expressed p16ink4a. In all, 1/7 SCC with undifferentiated precursors showed LVS invasion.
Stages of primary SCC at initial presentation were equally distributed in all 3 groups of precursors. There was a slightly higher percentage of LVS invasion and LN metastases in d-PeIN, however, without statistical significance. Similarly, higher stages of primary SCC were more prominent in d-PeIN, since LVS invasion and LN metastases correlated with stages.
Correlation of Peritumoral Precursor Lesions With Mutational Status of the Invasive SCCMore than half of the invasive SCC (37/70; 54%) harbored mutations in the tumor suppressor gene TP53 (34/37 SCC with at least 1 missense mutation), while 33/70 (46%) invasive SCC were devoid of TP53 hotspot mutations. Overall, 32/70 (46%) SCC featured CDKN2A mutations (4 missense and 28 nonsense or frameshift mutations). Other genes with recurrent mutations included HRAS (13/70 SCC; 19%), PIK3CA (13/70 SCC; 19%), FBXW7 (7/70 SCC; 10%) and FGFR3 (4/70 SCC; 6%; for details, see Fig. 2). Overall, 24/32 (75%) of CDKN2A mutated SCC showed a co-segregation with TP53, predominantly in SCC associated with d-PeIN (P=0.003). Oncogenic activating mutations in HRAS and PIK3CA occurred predominantly in the group of verrucous/verruciform invasive SCC with corresponding verrucous/verruciform precursor lesions (n=14; P=0.02; for details, see Fig. 2, for summary, see Tables 1, 2, Fig. 2).
Precancers Adjacent to SCC Carrying TP53 and CDKN2A Hotspot MutationsInvasive SCC with TP53 mutations and/or CDKN2A mutations featured d-PeIN (37/42; P<0.001) with variously elongated rete ridges and near-normal cornification, verrucous (4/37) and basaloid (1/37) precursors. All but 4 d-PeIN with TP53 missense mutations in the associated invasive SCC showed immunohistochemical nuclear p53 overexpression in basal and suprabasal keratinocytes of the peritumoral precursors (Figs. 1B, D, F). Null p53 staining in 1 d-PeIN correlated with a frameshift TP53 mutation in the associated SCC. One verrucous precursor harbored 2 TP53 hotspot mutations at a low mutational frequency of <25% each, which correlated with an inconsistent and focal p53 nuclear overexpression in both invasive SCC and precursor lesion (Figs. 3D–G). p16ink4a was overexpressed in 6 invasive SCC with missense CDKN2A mutation but in only 5 associated peritumoral d-PeIN (Figs. 4A–F). A lack of p16ink4a overexpression or patchy and weak staining in SCC and associated precursors correlated with nonsense or frameshift mutations in CDKN2A (Fig. 5). Two SCCs with an undifferentiated peritumoral precursor lesion harbored TP53 mutations, but only 1 SCC and associated precursor showed overexpression of p53. (Figs. 4C–F) One basaloid precursor lesion with p53 overexpression also had a concomitant CDKN2A missense mutations with p16ink4a overexpression.
 FIGURE 5:
FIGURE 5: Differentiated HPV-independent penile precursors (d-PeIN) with missense mutations in both TP53 and CDKN2A genes. A–D, pT2 SCC, missense mutations in TP53 (R175H) and CDKN2A (P81L). A, Hematoxylin and eosin–stained section: Overview showing the transition of invasive SCC, preinvasive precursor lesion, and lichen planus. B, Nuclear p53 overexpression in the invasive tumor cells and basal and suprabasal cells of the peritumoral precursor lesion. C, Cytoplasmic p16ink4a overexpression in the invasive tumor cells and basal cells of the peritumoral precursor lesion. D, High-power view of nuclear p53 and cytoplasmic p16ink4a staining in d-PeIN. E–H, pT2 SCC; missense mutation in TP53 (P278S) and CDKN2A (P114L). Hematoxylin and eosin–stained section: Overview showing the transition of preinvasive precursor lesion to keratinizing invasive SCC arising in chronic lichenoid inflammatory skin disease (E) with nuclear p53 overexpression in the invasive tumor cells and the peritumoral precursor lesion (F). G, The corresponding immunohistochemical stain shows cytoplasmic p16ink4a overexpression in the invasive tumor cells only. The d-PeIN is negative. High-power view of nuclear p53 overexpression and negative p16ink4a stain in d-PeIN (H).
Precancers of SCC Without TP53 Hotspot MutationsOf the 33 SCC without hotspot TP53 mutations, 18/33 (55%) SCC were flanked by verrucous hyperkeratotic precursors with wild-type p53 protein (P<0.001), 10/33 (30%) SCC by d-PeIN (4/10 with p53 overexpression, 6/10 wild-type p53 protein), 5/33 (15%) SCC by basaloid precursors (1/5 with p53 overexpression, 4/5 with p53 wild-type; and 2/15 (13%) by verruciform nonkeratinized precursor lesions; Figs. 4A,B). Five of 7 SCC with undifferentiated precursors were devoid of TP53 and CDKN2A hotspot mutations and lacked p16ink4a staining, but 1/5 showed nuclear p53 overexpression. Nuclear p53 overexpression in the absence of hotspot TP53 mutations (Figs. 1C, D) was interpreted as result of TP53 missense mutations not covered by the hotspot cancer panel.
DISCUSSIONTo the best of our knowledge, we present the first proposal of a simple classification of HPV-negative penile intraepithelial precursor lesions based on analysis of a cohort of 70 patients with HPV-negative penile SCC. We identified 4 characteristic histologic patterns, namely d-PeIN in the majority of cases, followed by verrucous and verruciform precursors, and the rare basaloid precursors. Histology was dependent on the site where the SCC arose, e.g. cornified mucosa or noncornified glycogenated mucosa.24,26,35 Almost 2/3 of precursor lesions were d-PeIN with the characteristic morphology with elongated rete ridges with basal cell atypia, premature squamatisation, and near-normal squamous maturation/cornification, and arose in the background of chronic lichenoid dermatoses. Less than one third of precursors were verrucous wild-type p53 precursor lesions with parakeratosis and hyperkeratosis, minimal cytologic atypia in plump rete ridges. They were reminiscent of vulvar precursor lesions described as vulvar acanthosis with altered differentiation.24,34,35 Noncornified verruciform precursors of invasive SCC arising in glycogenated mucosa were reminiscent of those described in mucosal sites of vulvar vestibulum or cervix.26 Verrucous and verruciform precursors were not associated with chronic inflammatory dermatoses. They correlated strongly with verrucous and papillary histology of invasive SCC, while the d-PeIN were observed with several histologies of invasive SCC. The undifferentiated 16ink4a negative HPV-independent penile intraepithelial lesions were the rarest subtype with 11%, compared with 19% in the original description.27,29 Interestingly, 10% of our initial cohort of HPV-negative SCC had no peritumoral precursor lesions but rather very short segments of hyperplasia of <3 rete ridges were identified, similar to observations in vulvar SCC.36
d-PeIN were identified adjacent to SCC featuring mutations in tumor suppressor genes TP53 and CDKN2A, while the SCC of verrucous/verruciform precursors lacked tumor suppressor gene mutations but harbored activating oncogenic mutation, mainly in PIK3CA and HRAS. Missense TP53 mutations in d-PeIN correlated with p53 overexpression while truncating mutations were associated with a null pattern. Rare d-PeIN with CDKN2A missense mutations showed p16ink4a overexpression, while d-PeIN with truncating or no CDKN2A mutations were p16ink4a negative. TP53 and CDKN2A mutations are present in early invasive HPV-negative penile SCC19 and in vulvar (pre)cancers.36,37 Overexpression of p53 and p16ink4a in penile precursors suggests an emergence already during the preinvasive stage of penile carcinogenesis. Both, verrucous and verruciform precursors, showed a p53 wild-type staining pattern. The small group of basaloid precursors was heterogeneous with respect to gene mutations and p53/p16ink4a overexpression. Pairing histology with the p53/p16ink4a profile of the penile precancers with correlation of the mutational profile of the invasive SCC, we identified at least 2 distinct pathways of carcinogenesis with 2 distinct highly differentiated precursor lesions, mirroring observations in HPV-negative vulvar24,35 and cervical precancers.26
Results from previous small cohort publications on penile HPV-independent precursors are difficult to compare. Some investigated mostly morphology with occasional p16ink4a staining.9,38,39 p53 was not excluded as a standard marker.9,40 Other authors proposed an antibody panel for workup but did not follow-up with a subclassification.23 For everyday surgical pathology practice, workup with antibodies to Ki-67, p16ink4a, and p53 should suffice for a correct classification of the vast majority of precancerous lesion. A continuous linear nuclear p53 overexpression in basal and suprabasal cells is typical for HPV-independent d-PeIN and differentiated vulvar intraepithelial neoplasia.41 An increased, albeit still discontinuous wild-type p53 staining pattern as result of ischemic and inflammatory stress in basal keratinocytes in hypertrophic dermatoses should not be overinterpreted as d-PeIN.36,42 In d-PeIN with p53 null pattern, the diagnostic clues of typical morphology, nuclear atypia, and mitotic activity should suffice for a correct diagnosis. The antibody panel is less valuable for unequivocal separation of verrucous HPV-independent penile precursors from benign reactive squamous proliferations or hypertrophic chronic inflammatory dermatoses. Abnormal maturation and cornification may assist in correctly diagnosing verrucous precursors, as prominent parakeratosis is neither a feature of a lichenoid dermatosis nor a benign squamous hyperplasia. A diagnostic dilemma represents the HPV-independent undifferentiated full-thickness precursor lesion,29 but lack of p16ink4a overexpression and optimally concomitant p53 overexpression allows correct identification of these rare precursors.29 Caution is advised with interpreting p16ink4a staining, as strong cytoplasmic overexpression of p16ink4a can also occur in the absence of a transforming HPV infection as a reflection of altered retinoblastoma pathways due to CDKN2A missense mutations. Patchy, heterogeneous, and weak staining can occur with truncating CDKN2A mutation. Overall, one should keep in mind, that not all histologically typical d-PeIN over express p53, that some rare HPV-negative precursor lesions may be p16ink4a positive, and seemingly benign verrucous and verruciform proliferations are true precursor lesions.
The type of precursor lesion impacts on therapy and prognosis, although no publications on the natural history of HPV-negative penile precursor lesions exist. Many assumptions are drawn from experience with vulvar carcinogenesis. Differentiated vulvar intraepithelial neoplasia can arise multifocally in chronic dermatoses and are classified as aggressive high-grade lesions with a rapid progression to invasive SCC, sometimes in several months or less than a year.43–45 In analogy, d-PeIN should be considered rapidly progressing precursors requiring immediate attention. The assumption of rapid progression is supported by observations that HPV-independent SCC are higher stage at primary initial presentation,19 and that penile carcinogenesis is associated with a poorer prognosis when compared with HPV-induced penile SCC.4 Furthermore, we observed a trend that SCC arising via d-PeIN had a slightly higher rate of LVS invasion and LN metastasis that correlated with higher stages of primary SCC at the time of diagnosis. The preferred therapy of all precursor subtypes is surgical resection, rather than destruction or medical therapy. Follow-up and prognosis depend on the type of precursor. Experiences from vulvar studies show that patients with residual dermatoses have a high risk for recurrence.46 Therapy of the (residual) dermatoses is necessary as it lowers the risk of recurrent/de novo carcinogenesis by reducing the inflammatory infiltrate and tissue damage.47 Data on verrucous and basaloid precursors are less abundant, but from personal experience, verrucous precursor lesions are often solitary and less likely to recur.
Overall, our data suggest at least 2 different genetic pathways for the development of HPV-independent penile SCC. One is associated with mutations in tumor suppressor genes in the background of chronic inflammatory skin diseases via the precursor lesion d-PeIN. The other pathway involves activating oncogenic mutations in proto-oncogenes rather than tumor suppressor gene mutations and gives rise to mainly verrucous and verruciform papillary invasive SCC via verrucous and verruciform precancers. Since carcinogenesis in various anogenital sites follows a universal pattern, it would seem only logical that the classification of HPV-independent penile precursor lesions follows the already established subtyping in vulva and cervix. Introducing separate classifications for penile HPV-independent precursors such as grade I-III d-PeIN, or new terms such as classic d-PeIN, hyperplasia-like d-PeIN (for verrucous precursors), and pleomorphic d-PeIN (for undifferentiated basaloid precursors)39 leads only to confusion among pathologists and oncologists, who deal with cancers of various different anogenital locations. Therefore, we propose a simple classification of HPV-independent penile precursor lesions with 2 major genetic/biological pathways requiring different clinical management decisions. These include the precursor
Comments (0)