
Remember me
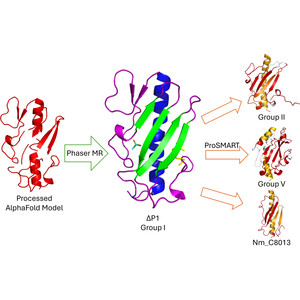
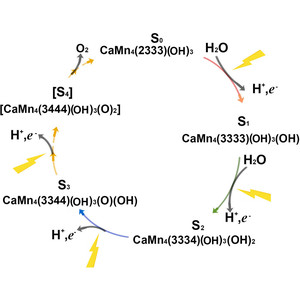

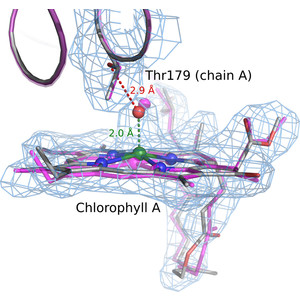







Model building and refinement, and the validation of their correctness, are very effective and reliable at local resolutions better than about 2.5 Å for both crystallography and cryo-EM. However, at local resolutions worse than 2.5 Å both the procedures and their validation break down and do not ensure reliably correct models. This is because in the broad density at lower resolution, critical features such as protein backbone carbonyl O atoms are not just less accurate but are not seen at all, and so peptide orientations are frequently wrongly fitted by 90–180°. This puts both backbone and side chains into the wrong local energy minimum, and they are then worsened rather than improved by further refinement into a valid but incorrect rotamer or Ramachandran region. On the positive side, new tools are being developed to locate this type of pernicious error in PDB depositions, such as CaBLAM, EMRinger, Pperp diagnosis of ribose puckers, and peptide flips in PDB-REDO, while interactive modeling in Coot or ISOLDE can help to fix many of them. Another positive trend is that artificial intelligence predictions such as those made by AlphaFold2 contribute additional evidence from large multiple sequence alignments, and in high-confidence parts they provide quite good starting models for loops, termini or whole domains with otherwise ambiguous density.
Comments (0)