
Remember me





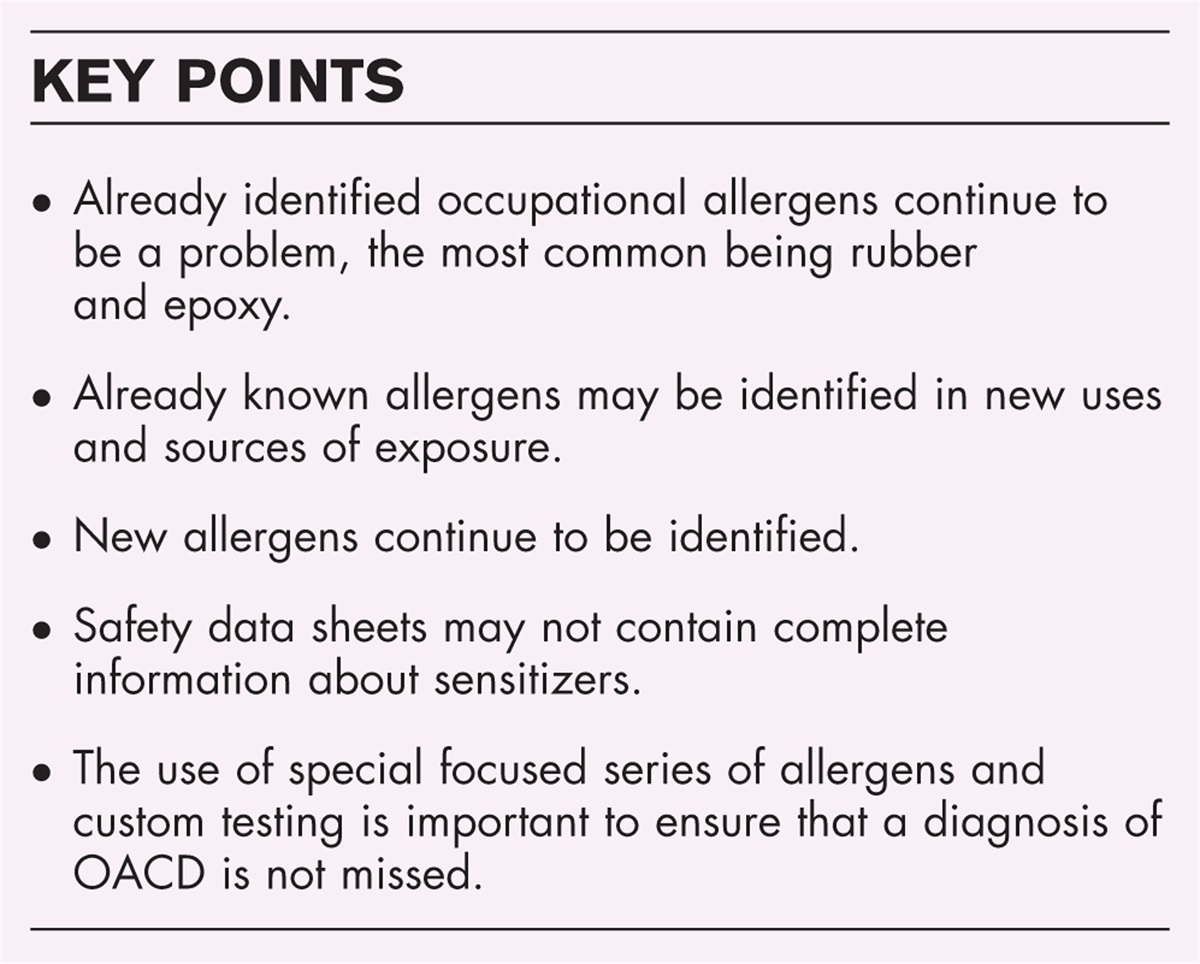


Common variable immunodeficiency (CVID) is the most prevalent symptomatic human inborn error of immunity (IEI), with a prevalence of 1 : 10 000–1 : 50 000 in Caucasians [1]. The clinical phenotype of affected patients is highly heterogeneous, encompassing increased susceptibility to infectious episodes, autoimmune phenomena, polyclonal lymphoproliferation, granulomatous disease, and increased risk in developing malignancy [2▪]. Rather than a single entity, CVID is considered as an umbrella term embracing an heterogenous group of clinical and immunological phenotypes, that may be caused by various genetic and/or environmental factors [2▪]. In fact, and conversely from other IEIs which are characterized by a specific genetic defect in only 10–30% of patients with a CVID reach a definite genetic diagnosis, while for the remaining patients the disease is considered polygenic/multifactorial rather than one based on mendelian inheritance [3,4]. Therapeutical strategies for CVID affected patients are mainly symptomatic, requiring life-long immunoglobulin replacement treatment (IgRT) as a mainstay; in addition, antimicrobial courses may be frequently required when infectious episodes occur, as well as immunosuppression/modulation for the management of autoimmune phenomena or chronic lymphoproliferation [5▪]. However, and with the exception of IgRT which reduces infectious episodes’ rates and severity, the natural history of noninfectious CVID-related complication is hardly predictable and empirical treatment is often adopted. Nonetheless, besides unveiling more insights on the human immune system biology, understanding the molecular basis of CVID and CVID-like disorders enables the physicians to exploit the molecular defect with targeted treatments. In this review, we will focus on monogenic forms of CVID for which a target treatment is currently available or under investigation.
 Box 1:
Box 1: no caption available
TEXT OF REVIEW Activated phosphoinositide 3-kinase delta syndromeClass I phosphatidylinositol-3 kinases (PI3Ks) sense and transduce external signals which are fundamental for cell metabolism, differentiation, proliferation, and survival [6]. PI3Ks are formed by heterodimers comprising a regulatory (p85α, p55α, p50α, p85β, or p55γ) and a catalytic (class IA p110α, β, or δ; class IB p110γ) subunit; while p110α and β are ubiquitously expressed, p110δ and γ are primarily expressed in the immune system [6].
Activated phosphoinositide 3-kinase δ syndrome (APDS) is a recently described IEI characterized by recurrent sinopulmonary infectious episodes, bronchiectasis, impaired viral clearance, chronic lymphoproliferation, autoimmunity, and increased lymphoma risk [7]. The disease, inherited with an autosomal dominant manner, is caused by heterozygous gain-of-function mutations on the catalytic p110δ (coded by the PIK3CD gene, APDS-1, OMIM #615513) or by heterozygous loss-of-function mutations on the regulatory p85α (coded by the PI3KR1 gene, APDS-2, OMIM #616005) subunit of the PI3K [8–11]. In both cases, a downstream hyperactivation of the PI3K pathway results in increased and sustained signaling on the mammalian/mechanistic target of rapamycin (mTOR) as well as c-Myc activation, which leads to metabolic reprogramming directed toward expansion and proliferation, ultimately resulting in an exhausted and senescent T-cell profile [12,13]. In addition, B cell signaling and maturation are also affected: impaired class-switch recombination and plasmablast maturation are observed, leading to poor humoral response, as well as loss of self-tolerance and survival of autoreactive immunoglobulin M (IgM)-secreting B cells, contributing to the development of autoimmune manifestations [14,15].
Since the first description of APDS in 2013, more than 250 patients have been described expanding the clinical and immunological features of the disease In fact, recurrent respiratory tract infectious episodes are the most common infectious manifestation in both APDS and CVID patients, as well as typical noninfectious complications such as autoimmune phenomena (i.e. enteropathy, autoimmune cytopenia, arthritis) or chronic benign lymphoproliferation (chronic adenopathies, hepatosplenomegaly, mucosal nodular lymphoid hyperplasia) [16▪,17,18]. On the contrary, additional nonimmunological features such as neurodevelopmental delay or neuropsychiatric disorders have been described in both APDS-1 and APDS-2 patients, manifestations, which are rather uncommon in CVID patients [18,19]. Peculiar immunological alterations that should raise suspicion for APDS are represented by raised IgM serum levels in a context of decreased IgG and IgA serum levels, progressive B cell lymphopenia with a relative expansion of transitional B cells and plasmablasts, decrease of naïve T cell subsets with relative expansion of central and effector memory T cells [16▪,18,20▪▪]. Nevertheless, taking in consideration the overlapping clinical phenotype of CVID and APDS patients, genetic screening including PIK3CD and PIK3R1 should be offered to all genetically undefined CVID patients. The actual prevalence of APDS-causing mutations among CVID patients is difficult to estimate, as genetic analysis in CVID patients is not worldwide uniformed; however, data from large cohorts’ genetic studies revealed that pathogenic PIK3CD or PIK3R1 mutations are found in approximately 1.4–7.1% of CVID patients [4,21,22]. Although displaying low genetic heterogeneity, APDS patients carrying the canonical E1021K mutation present highly variable clinical phenotype, thus suggesting a role for additional genetic/epigenetic or environmental factors in contributing to the disease manifestations [20▪▪]. The pathogenicity of novel unreported mutations must be confirmed by evaluating phosphorylation levels of AKT and/or S6 proteins in patients’ activated T or B lymphocytes [23].
Initially, treatment strategies for APDS patients resembled those adopted for CVID and were mainly symptomatic, including IgRT, antibiotic and/or antiviral treatment or prophylaxis, respiratory physiotherapy, and immunosuppressive drugs (e.g. corticosteroids, rituximab, azathioprine, mycophenolate, anti-TNFα monoclonal antibodies) [17,24,25]. However, and consistent with the hyperactivated mTOR pathway, a first attempt of tailored medicine was represented by the use of rapamycin: while highly effective on controlling chronic lymphoproliferation, it showed less robust results on cytopenias and colitis, even though with an adequate safety profile [17]. Hematopoietic stem cell transplantation (HSCT) represents the only curative approach for APDS; however, even though the results in terms of overall survival (OS) are satisfactory (86% 2-year OS), graft instability consequentially leading to graft failure requiring unplanned donor cell infusion represented a major limitation to successful HSCT [21,26▪,27▪▪]. Therefore, conservative treatment with the possibility of target therapy seems, at least for now, much more encouraging.
Recently, the results of a randomized, placebo-controlled, phase 3 trial of the PI3Kδ inhibitor Leniolisib have been published [28▪▪]. Leniolisib was administered to APDS patients aged 12 years or older at a dose of 70 mg twice daily over a period of 12 weeks; the drug was well tolerated with more drug-related adverse events in the placebo group rather than the Leniolisib one. Compared to placebo, Leniolisib effectively controlled chronic polyclonal lymphoproliferation, reducing lymph nodes as well as spleen size. Amelioration of immunological parameters such as increase of naïve B cell, decrease of serum IgM and improvement of autoimmune cytopenias also occurred. Finally, expanded transitional B cells and raised senescent CD8+ T cells, which are both thought to contribute to defective viral clearance, were both improved [28▪▪]. In March 2023, Leniolisib was approved in the United States by the Food and Drug Administration (FDA) for the treatment of APDS in adult or pediatric patients older than 12 years and is currently also under regulatory review in the European Union [29]. Besides Leniolisib, other PI3Kδ inhibitors have been investigated for APDS, with less promising results. An open-label trial of the inhaled agent Nemiralisib was completed in 5 APDS patients: even though Nemiralisib was well tolerated, the trial did not provide evidence regarding its efficacy in target engagement in the lung, as well as downstream effects modulating local lung or systemic blood inflammation, which could have been of benefit for APDS-affected patients [30].
Cytotoxic T lymphocyte antigen 4 haploinsufficiency and lipopolysaccharide-responsive beige-like anchor protein deficiencyCytotoxic T lymphocyte antigen 4 (CTLA-4) is a key T cell co-receptor which acts as a negative regulator in maintaining immune homeostasis by downregulating CD28:B7 ligands (CD80/CD86) interactions [31]. CTLA-4 plays its regulatory function both in a cell-extrinsic manner, where T-regulatory (T-reg) cells downregulate B7 by trans-endocytosis and degradation, and in a cell-intrinsic manner, by limiting B7 availability on the surface of T cells via cis-endocytosis [32▪,33]. CTLA-4 haploinsufficiency causes a severe CVID-like monogenic IEI with predominantly immune dysregulatory features characterized by progressive B cell exhaustion and hypogammaglobulinemia, multiorgan autoimmunity (immune cytopenia, enteropathy, endocrinopathies), and chronic lymphoproliferation with lymphocytic infiltrates in several organs (brain, gut, liver, and lung) [34,35]. CTLA-4 mutated patients express a highly heterogeneous clinical phenotype and incomplete disease penetrance: additional genetic or epigenetic, as well as environmental factors, are suspected to contribute to the clinical picture, even though this is still under investigation. Common infectious agents [such as Epstein−Barr virus (EBV) or cytomegalovirus (CMV)] were thought to act as a trigger; however, a recent study investigated the seroprevalence of EBV, CMV, Herpes simplex 1/2, Parvovirus B19 and Toxoplasma gondii among affected and unaffected CTLA-4 mutated subjects, finding no differences [36▪].
Patients with biallelic loss-of-function mutations in the lipopolysaccharide-responsive beige-like anchor protein (LRBA) gene are clinical phenocopies of CTLA-4 haploinsufficiency [37]. From a molecular point of view, LRBA prevents AP-1 driven CTLA-4 lysosomal degradation by recycling and cell surface transferring CTLA-4-containing vesicles; therefore, patients lacking wild-type LRBA expression display decreased CTLA-4 expression and altered T-reg function, thus causing immune dysregulation and autoimmunity [38]. Compared to CTLA-4 mutated patients, LRBA deficiency affected patients tend to present a more severe clinical picture with earlier disease onset; the clinical picture comprises recurrent or invasive infectious episodes, enteropathy, autoimmune cytopenia, granulomatous disease, lymphoproliferation [39,40,41▪]. Immunological analysis revealed normal T cell count with reduced T-regs as well as B cells perturbations such as reduced switched-memory B cells and plasmablasts and expanded autoreactive CD21lo B cells [39,40].
Even though both disorders affect the expression and/or function of the same immunological check-point inhibitor, LRBA deficient patients tend to present a more severe phenotype and a reduced OS – one possible explanation is given by a variable disease penetrance reported for CTLA-4 mutated patients, whereas patients with LRBA deficiency normally present with complete penetrance and fully overt disease presentation [35,41▪]. Data from single-center or nationwide study revealed that the prevalence of pathogenic CTLA-4 or LRBA mutations among subjects with a CVID/CVID-like diagnosis ranges between 1.7–20.8% and 0.9–7.2%, respectively [4,22]. Taking this into account, CTLA-4 haploinsufficiency and LRBA deficiency represent two of the most common causes of monogenic CVID, therefore suggesting the vital importance of screening CVID patients for these two genes, both in terms of proper follow-up and due to the possibility of establishing a tailored treatment [4,42,43].
Besides IgRT for those patients presenting with hypogammaglobulinemia, additional treatment choices for patients with CTLA-4 haploinsufficiency or LRBA deficiency are mainly directed towards autoimmune manifestations and/or organ lymphocytic infiltrates [39,44▪▪]. Conventional immunosuppressive drugs (systemic steroids, Mycophenolate Mophetile, Azathioprine) or T-cell modulators (Cyclosporine, Cyclophosphamide) were extensively used either alone or in combination to control autoimmune manifestations such as enteropathy or autoimmune cytopenias; in addition, the use of the mTOR inhibitor Sirolimus may enhance T-reg response [39,44▪▪]. B lymphocyte depleting therapy with anti-CD20 monoclonal antibodies showed good results in treating lymphoid infiltration, as B cells express CD80/CD86 and therefore T-cell activation can be limited [39,44▪▪].
Abatacept is a soluble fusion protein composed of the Fc IgG1 region and the CTLA-4 extracellular domain that selectively binds CD80/CD86, thus inhibiting the activation of CD28+ T cells [45,46]. Based on its biological function and with the rationale of providing exogenous CTLA-4 molecule, Abatacept has been implemented in several CTLA-4 or LRBA mutated patients: dosing scheme for adult patients usually start with a loading dose of 500–1000 mg intravenously, followed then by 125–250 mg per week subcutaneously [44▪▪,47,48,49▪]. In pediatric patients with CTLA-4/LRBA-related disorders, Abatacept has be administered at a dose of 10–20 mg/kg every 2 weeks until complete remission, without major adverse events [49▪,50]. Even though Abatacept is reported to show high efficacy in controlling chronic enteropathy and lymphoproliferation, variable results with partial remission were seen in other immunedisregulatory features such as autoimmune cytopenia or neurological manifestations [44▪▪,48]. By contrast, in CTLA-4 mutated patients with granulomatous lymphocytic lung disease the use of Abatacept could lead to full or partial resolution in up to 70% of the treated patients [44▪▪]. As evidence of Abatacept use in CTLA-4/LRBA mutated patients is mainly derived from retrospective studies or case reports, a phase IIa prospective, nonrandomized, open-label, single arm multicenter trial (ABACHAI trial) is currently evaluating the safety and efficacy of Abatacept in patients with CTLA-4 insufficiency or LRBA deficiency and results are expected to be published in 2023 [51▪].
Besides the promising results and the possibility of target treatments, HSCT still represents the only curative approach for both disorders; nevertheless, reported patients are scarce and with variable results [52,53]. Currently, recommendations on HSCT for these diseases are lacking and indications for HSCT are mainly directed towards those patients presenting multiple organs involvement refractory to conventional treatment and/or severe invasive infectious episodes [39,44▪▪,52–54]. As improving organ function is vital for achieving better HSCT outcomes, patients with CTLA-4 haploinsufficiency or LRBA deficiency might benefit from the use of Abatacept as a bridge to HSCT, rather than as continuative chronic treatment. In addition, the optimal minimal chimerism sufficient for achieving clinical and immunological control of the disease is still unclear [55▪]. Finally, the results of a CRISPR-Cas9 gene editing for CTLA-4 haploinsufficiency have been recently published: the adopted approach led to CTLA-4 protein restoration and effective CD80/CD86 transendocytosis both in in vitro patients’ T cells and in vivo ctla-4−/− mouse model, opening the possibility for a safer yet equally effective curative option than HSCT [56▪▪].
CONCLUSIONIn conclusion, CVID/CVID-like disorders present with heterogenous clinical and immunological manifestations, that may be caused by various genetic or environmental factors. In this review, we focalized our attention on three different monogenic forms of CVID-like disease and described new targeted strategies implicated in the different altered immunological pathways. The first one involves the PI3K pathway causing activated phosphoinositide 3-kinase delta syndrome, for which a direct oral inhibitor has been recently approved by the FDA. The other two involve T-regs maturation and homeostasis and are caused by defects in CTLA-4 or LRBA gene; a soluble costimulation modulator (Abatacept) has been widely used showing good results in disease control. Targeted treatments in these disorders allow for significant control of noninfectious disease related complications, thus setting the basis for ameliorating prognosis and quality of life of affected patients. Furthermore, the costs and availability of these drugs may become a limitation depending on factors such as country of residence, type of national health system and others, especially since national and international guidelines for treatment of affected patients are not well defined yet. Understanding the biological mechanism of monogenic forms of CVID disorders may lead to the development of targeted therapies allowing for the application of precision medicine in the setting of IEIs, with evident implications for patients’ prognosis and quality of life.
AcknowledgementsWe would like to thank the patient, the patient's family and the nurses for all their efforts. Several of us are members of the European Reference Network for Rare Immunodeficiency, Autoinflammatory and Autoimmune Diseases (project identification no. 739543).
Financial support and sponsorshipThe research leading to these results has received funding from the European Community's Seventh Framework Programme FP7/2007–2013 under grant agreement no 201549 (EURO-PADnet HEALTH-F2-2008-201549) and from the Italian Ministerial Grant GR-2010-2315762. The research leading to these results also received funding from the ‘Fondazione C. Golgi’, Brescia, Italy and the Jeffrey Modell Foundation.
Conflicts of interestThere are no conflicts of interest.
REFERENCES AND RECOMMENDED READINGPapers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES 1. Bonilla FA, Barlan I, Chapel H, et al. International Consensus Document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract 2016; 4:38–59. 2▪. Tangye SG, Al-Herz W, Aziz B, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 2022; 42:1508–1520. 3. Ramirez NJ, Posadas-Cantera S, Caballero-Oteyza A, et al. There is no gene for CVID—novel monogenetic causes for primary antibody deficiency. Curr Opin Immunol 2021; 72:176–185. 4. Rojas-Restrepo J, Caballero-Oteyza A, Huebscher K, et al. Establishing the molecular diagnoses in a cohort of 291 patients with predominantly antibody deficiency by targeted next-generation sequencing: experience from a monocentric study. Front Immunol 2021; 12:786516. 5▪. Fevang B. Treatment of inflammatory complications in common variable immunodeficiency (CVID): current concepts and future perspectives. Expert Rev Clin Immunol 2023; 19:627–638. 6. Rathinaswamy MK, Burke JE. Class I phosphoinositide 3-kinase (PI3K) regulatory subunits and their roles in signaling and disease. Adv Biol Regul 2020; 75:100657. 7. Coulter TI, Chandra A, Bacon CM, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: a large patient cohort study. J Allergy Clin Immunol 2017; 139:597–606. e4. 8. Lucas CL, Kuehn HS, Zhao F, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol 2014; 15:88–97. 9. Angulo I, Vadas O, Garçon F, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science 2013; 342:866–871. 10. Deau MC, Heurtier L, Frange P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest 2014; 124:3923–3928. 11. Lucas CL, Zhang Y, Venida A, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med 2014; 211:2537–2547. 12. Cannons JL, Villarino AV, Kapnick SM, et al. PI3Kδ coordinates transcriptional, chromatin, and metabolic changes to promote effector CD8+ T cells at the expense of central memory. Cell Rep 2021; 37:109804. 13. Bier J, Rao G, Payne K, et al. Activating mutations in PIK3CD disrupt the differentiation and function of human and murine CD4+ T cells. J Allergy Clin Immunol 2019; 144:236–253. 14. Avery DT, Kane A, Nguyen T, et al. Germline-activating mutations in PIK3CD compromise B cell development and function. J Exp Med 2018; 215:2073–2095. 15. Lau A, Avery DT, Jackson K, et al. Activated PI3Kδ breaches multiple B cell tolerance checkpoints and causes autoantibody production. J Exp Med 2020; 217:e20191336. 16▪. Qiu L, Wang Y, Tang W, et al. Activated phosphoinositide 3-kinase δ syndrome: a large pediatric cohort from a single center in China. J Clin Immunol 2022; 42:837–850. 17. Maccari ME, Abolhassani H, Aghamohammadi A, et al. Disease evolution and response to rapamycin in activated phosphoinositide 3-kinase δ syndrome: The European society for immunodeficiencies-activated phosphoinositide 3-kinase δ syndrome registry. Front Immunol 2018; 9:543. 18. Oh J, Garabedian E, Fuleihan R, Cunningham-Rundles C. Clinical manifestations and outcomes of activated phosphoinositide 3-kinase δ syndrome from the USIDNET Cohort. J Allergy Clin Immunol Pract 2021; 9:4095–4102. 19. Elkaim E, Neven B, Bruneau J, et al. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase δ syndrome 2: a cohort study. J Allergy Clin Immunol 2016; 138:210–218. e9. 20▪▪. Maccari ME, Wolkewitz M, Schwab C, et al. Activated phosphoinositide 3-kinase δ syndrome: update from the ESID Registry and comparison with other autoimmune-lymphoproliferative inborn errors of immunity. J Allergy Clin Immunol 2023; doi: 10.1016/j.jaci.2023.06.015. [Epub ahead of print]. 21. Okano T, Imai K, Tsujita Y, et al. Hematopoietic stem cell transplantation for progressive combined immunodeficiency and lymphoproliferation in patients with activated phosphatidylinositol-3-OH kinase δ syndrome type 1. J Allergy Clin Immunol 2019; 143:266–275. 22. Abolhassani H, Hammarström L, Cunningham-Rundles C. Current genetic landscape in common variable immune deficiency. Blood 2020; 135:656–667. 23. Moriya K, Mitsui-Sekinaka K, Sekinaka Y, et al. Clinical practice guideline for activated phosphatidyl inositol 3-kinase-delta syndrome in Japan. Immunol Med 2023; 1–5. doi: 10.1080/25785826.2023.2210366. [Epub ahead of print]. 24. Tessarin G, Rossi S, Baronio M, et al. Activated phosphoinositide 3-kinase delta syndrome 1: clinical and immunological data from an italian cohort of patients. J Clin Med 2020; 9:3335. 25. Jamee M, Moniri S, Zaki-Dizaji M, et al. Clinical, immunological, and genetic features in patients with activated PI3Kδ syndrome (APDS): a systematic review. Clin Rev Allergy Immunol 2020; 59:323–333. 26▪. Yang X, Xi R, Bai J, Pan Y. Successful haploidentical hematopoietic stem cell transplantation for activated phosphoinositide 3-kinase δ syndrome: case report and literature review. Medicine 2023; 102:e32816. 27▪▪. Dimitrova D, Nademi Z, Maccari ME, et al. International retrospective study of allogeneic hematopoietic cell transplantation for activated PI3K-delta syndrome. J Allergy Clin Immunol 2022; 149:410–421. e7. 28▪▪. Rao VK, Webster S, Šedivá A, et al. A randomized, placebo-controlled phase 3 trial of the PI3Kδ inhibitor leniolisib for activated PI3Kδ syndrome. Blood 2023; 141:971–983. 29. Duggan S, Al-Salama ZT. Leniolisib: first approval. Drugs 2023; 83:943–948. 30. Begg M, Amour A, Jarvis E, et al. An open label trial of nemiralisib, an inhaled PI3 kinase delta inhibitor for the treatment of Activated PI3 kinase Delta Syndrome. Pulm Pharmacol Ther 2023; 79:102201. 31. Gámez-Díaz L, Seidel MG. Different apples, same tree: visualizing current biological and clinical insights into CTLA-4 insufficiency and LRBA and DEF6 deficiencies. Front Pediatr 2021; 9:662645. 32▪. Xu X, Dennett P, Zhang J, et al. CTLA4 depletes T cell endogenous and trogocytosed B7 ligands via cis-endocytosis. J Exp Med 2023; 220:e20221391. 33. Wei SC, Sharma R, Anang NAAS, et al. Negative co-stimulation constrains T cell differentiation by imposing boundaries on possible cell states. Immunity 2019; 50:1084–1098. e10. 34. Kuehn HS, Ouyang W, Lo B, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 2014; 345:1623–1627. 35. Schwab C, Gabrysch A, Olbrich P, et al. Phenotype, penetrance, and treatment of 133 CTLA-4-insufficient subjects. J Allergy Clin Immunol 2018; 142:1932. 36▪. Krausz M, Mitsuiki N, Falcone V, et al. Do common infections trigger disease-onset or -severity in CTLA-4 insufficiency? Front Immunol 2022; 13:1011646. 37. Lopez-Herrera G, Tampella G, Pan-Hammarström Q, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet 2012; 90:986–1001. 38. Gámez-Díaz L, Grimbacher B. Immune checkpoint deficiencies and autoimmune lymphoproliferative syndromes. Biomed J 2021; 44:400–411. 39. Tesch VK, Abolhassani H, Shadur B, et al. Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. J Allergy Clin Immunol 2020; 145:1452–1463. 40. Habibi S, Zaki-Dizaji M, Rafiemanesh H, et al. Clinical, immunologic, and molecular spectrum of patients with LPS-responsive beige-like anchor protein deficiency: a systematic review. J Allergy Clin Immunol Pract 2019; 7:2379–2386. e5. 41▪. Catak MC, Akcam B, Bilgic Eltan S, et al. Comparing the levels of CTLA-4-dependent biological defects in patients with LRBA deficiency and CTLA-4 insufficiency. Allergy 2022; 77:3108–3123. 42. Bruns L, Panagiota V, von Hardenberg S, et al. Common variable immunodeficiency-associated cancers: the role of clinical phenotypes, immunological and genetic factors. Front Immunol 2022; 13:742530. 43. de Valles-Ibáñez G, Esteve-Solé A, Piquer M, et al. Evaluating the genetics of common variable immunodeficiency: monogenetic model and beyond. Front Immunol 2018; 9:636. 44▪▪. Egg D, Rump IC, Mitsuiki N, Rojas-Restrepo J, et al. Therapeutic options for CTLA-4 insufficiency. J Allergy Clin Immunol 2022; 149:736–746. 46. Lorenzetti R, Janowska I, Smulski CR, et al. Abatacept modulates CD80 and CD86 expression and memory formation in human B-cells. J Autoimmun 2019; 101:145–152. 47. Lo B, Zhang K, Lu W, et al. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 2015; 349:436–440. 48. Kiykim A, Ogulur I, Dursun E, et al. Abatacept as a long-term targeted therapy for LRBA deficiency. J Allergy Clin Immunol Pract 2019; 7:2790–2800. e15. 49▪. Dhunputh C, Ducassou S, Fernandes H, et al. Abatacept is useful in autoimmune cytopenia with immunopathologic manifestations caused by CTLA-4 defects. Blood 2022; 139:300–304. 50. Lanz AL, Riester M, Peters P, et al. Abatacept for treatment-refractory pediatric CTLA4-haploinsufficiency. Clin Immunol 2021; 229:108779. 51▪. Krausz M, Uhlmann A, Rump IC, et al. The ABACHAI clinical trial protocol: Safety and efficacy of abatacept (s.c.) in patients with CTLA-4 insufficiency or LRBA deficiency: a non controlled phase 2 clinical trial. Contemp Clin Trials Commun 2022; 30:101008. 52. Seidel MG, Böhm K, Dogu F, et al. Treatment of severe forms of LPS-responsive beige-like anchor protein deficiency with allogeneic hematopoietic stem cell transplantation. J Allergy Clin Immunol 2018; 141:770–775. e1. 53. Slatter MA, Engelhardt KR, Burroughs LM, et al. Hematopoietic stem cell transplantation for CTLA4 deficiency. J Allergy Clin Immunol 2016; 138:615–619. e1. 54. Lougaris V, Malagola M, Baronio M, et al. Successful hematopoietic stem cell transplantation for complete CTLA-4 haploinsufficiency due to a de novo monoallelic 2q33.2-2q33.3 deletion. Clin Immunol 2020; 220:108589. 55▪. Margarit-Soler A, Deyà-Martínez À, Canizales JT, et al. Challenges in immune reconstitution following hematopoietic stem cell transplantation for CTLA-4 insufficiency-like primary immune regulatory disorders. Front Immunol 2022; 13:1070068. 56▪▪. Fox TA, Houghton BC, Petersone L, et al. Therapeutic gene editing of T cells to correct CTLA-4 insufficiency. Sci Transl Med 2022; 14:668.
Comments (0)