2.1 Ethics
The clinical study described herein and the informed consent form was approved by an Institutional Review Board (IRB) at the investigational site before enrollment of any subjects into the study and was conducted in accordance with the International Conference on Harmonization Good Clinical Practice guidelines and with the ethical principles of the Declaration of Helsinki. This study was conducted at a single site located in Daytona Beach, FL, USA (Covance).
2.2 Subjects
Eligible subjects were males or females aged 18–50 years; a body mass index of 18.0–32.0 kg/m2; normal or intermediate CYP2D6 metabolizers based on genetic testing; and in generally good health based on screening physical examination, medical history, 12-lead electrocardiogram (ECG), and clinical laboratory evaluations. Exclusion criteria included a history or current clinical manifestation of any clinically significant disorder or drug allergies; pregnant or lactating females; history of surgery potentially altering absorption and/or excretion; any prescription medications/products other than nonhormonal intrauterine devices within 14 days prior to check-in; or any medications/products known to alter drug absorption, metabolism, or elimination processes, including St. John’s Wort, within 35 days prior to dosing.
2.3 Study Design
This was a phase I, open-label, fixed sequence study to investigate the effect of paroxetine administration on the PK of ulotaront in healthy male and female subjects. Ulotaront exhibits dose-proportional PK in the dose range of 10–100 mg which covers its therapeutic dose range from 25 to 100 mg, and plasma exposure to ulotaront does not significantly accumulate after repeated daily dosing [3]. Therefore, to allow for the possibility of a significant increase in ulotaront plasma exposure under DDI conditions, single doses of 25 mg were selected for the study, with the first dose to be administered in the absence of paroxetine and the second dose in the presence of paroxetine. Twenty-four subjects were dosed to ensure that 20 subjects completed the study. The number of subjects planned provided 90% power to reject the null hypothesis that the ratio of test mean to reference mean was below 0.80 or above 1.25 for the primary endpoints, assuming that the expected ratio of the mean was within 5%, and the standard deviation (SD) difference (log scale) was 0.212, with one-sided alpha at the 5% level.
Subjects who discontinued the study or withdrew were not replaced. Subjects received a single dose of 25 mg ulotaront on Day 1 followed by paroxetine 20 mg once daily on Days 5–10 to achieve steady-state plasma paroxetine levels. On Day 11, subjects received a second single oral dose of 25 mg ulotaront with an oral dose of 20 mg paroxetine followed by 20 mg paroxetine once daily on Days 12–14. Subjects received ulotaront in the morning after a minimum 8-h overnight fast, and fasted for an additional 4 h post-dose of ulotaront.
2.4 Bioanalytical Methods and Assay Performance
Two validated curve-range bioanalytical methods with a liquid chromatography-tandem mass spectrometry (LC-MS/MS) were used for determination of ulotaront and its N-desmethyl metabolite SEP-363854 concentrations in plasma. The initial bioanalytical method with a lower limit of quantification (LLOQ) of 0.0200 ng/mL and a calibration range from 0.02 to 20 ng/mL for both ulotaront and SEP-363854 (using 200 μL plasma) was validated [6]; an extended curve range from 0.25 to 250 ng/mL (using 100 μL plasma) was then also validated to avoid a large portion of sample dilution needed in supporting clinical studies. During the PK sample measurement for this DDI study, samples were first analyzed by the high curve range method and those samples < 0.25 ng/mL were repeated using the low curve-range method. Thus, the LLOQ for this study was 0.02 ng/mL for both ulotaront and the metabolite. Overall, 19 analytical runs were processed, and the assay precision and accuracy of quality control (QC) samples were found to be 2.6–5.5% coefficient of variation (CV) and − 6.9 to 3.0% relative error (RE) for ulotaront, and 3.1–7.4% CV and − 8.8% to + 0.7% RE for the metabolite SEP-363854, respectively. To further confirm the assay robustness, about 10% of the total analyzed samples were selected for incurred sample reanalysis (ISR) reproducibility evaluation. The ISR results for both analytes had a 98% pass rate (within 20% difference). Both the inter-run precision and accuracy data and the ISR results demonstrated excellent assay performance.
For the period of dosing with paroxetine, plasma paroxetine concentrations were measured by a Covance-owned liquid-liquid extraction coupled with the LC-MS/MS method (paroxetine-d6 as IS) with an LLOQ of 0.0500 ng/mL and a validated curve range of 0.05–50 ng/mL using 100 μL of human plasma. Across 13 sample analysis runs for this study, precision and accuracy of QC samples were 1.6–5.4% CV and − 2.1% to 0.7% RE (i.e. 97.1–100.7% accuracy), respectively. The ISR results from about 10% of the total samples showed 100% meeting the acceptance criterion (within 20% difference), also indicating solid assay reproducibility.
2.5 Pharmacokinetic Analysis
Blood samples for determination of plasma ulotaront and SEP-363854 concentrations were collected on two series starting from Day 1 (to Day 5) and Day 11 (to Day 15) at predose (before study drug administration), as well at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 24, 48, 54, 72, 78, and 96 h post-dose. Day 1 series of 96 h post-dose and Day 11 series were also analyzed for plasma paroxetine concentrations. Trough blood samples from Day 6 to Day 10 at predose (before paroxetine administration) were also collected for determination of plasma paroxetine concentrations.
The PK parameters were determined by noncompartmental methods using Phoenix WinNonlin® version 8.1 (Certara USA, Inc., Princeton, NJ, USA) based on the individual plasma concentration-time data and actual sample collection times. The primary endpoints were area under the plasma concentration-time curve from time zero to infinity (AUC∞) and maximum observed plasma concentration (Cmax) for ulotaront on Day 1 (ulotaront alone) and Day 11 (ulotaront plus paroxetine). All other PK parameters were regarded as secondary or exploratory.
2.6 Pharmacogenomic Assessments
Subjects had a blood sample collected at screening for genetic polymorphism assessment of CYP2D6 using Luminex Platform (xTAG Luminex CYP2D6 IVD assay) by Covance Central Laboratory Services (Indianapolis, IN, USA). This genotyping assay solely detects variants *1(WT),*2, *3, *4, *5, *7, *8, *9, *10, *11, *15, *17, *29, *35, and *41. Definitions of normal metabolizer (NM), intermediate metabolizer, and PM follow Clinical Pharmacogenetics Implementation Consortium [22]. The genotyping and phenotype results for the subjects enrolled in this study are provided in electronic supplementary material [ESM] Table 1.
2.7 Statistical Analysis
For the primary analyses, only subjects with data for the given parameter for both treatments (ulotaront alone and ulotaront and paroxetine) were included. The primary analyses were conducted using a linear mixed-effects model, with the natural log-transformed PK parameter as the dependent variable, treatment as the fixed effect, and subject as a random effect. From this analysis, the point estimates and the corresponding two-sided 90% CIs and the treatment differences between ulotaront plus paroxetine and ulotaront alone, were calculated. These log-transformed results were back-transformed to the original scale by the exponentiation to obtain point estimates and the corresponding two-sided 90% CIs for the ratio of the geometric means of the primary PK parameters between the treatments of ulotaront plus paroxetine and ulotaront alone. All analyses were conducted using SAS version 9.4 or higher (SAS Institute, Inc., Cary, NC, USA).
2.8 Safety Assessments
Safety and tolerability were assessed by clinical laboratory tests, vital signs, ECGs, physical examinations, Columbia-Suicide Severity Rating Scale, and monitoring of AEs. AEs were monitored throughout the study at all visits. Clinical laboratory tests, vital signs, 12-lead ECGs, physical examination, and the Columbia-Suicide Severity Rating Scale were conducted throughout the study period. System organ class and preferred terms from the Medical Dictionary for Regulatory Activities (MedDRA, version 22.0) were used.
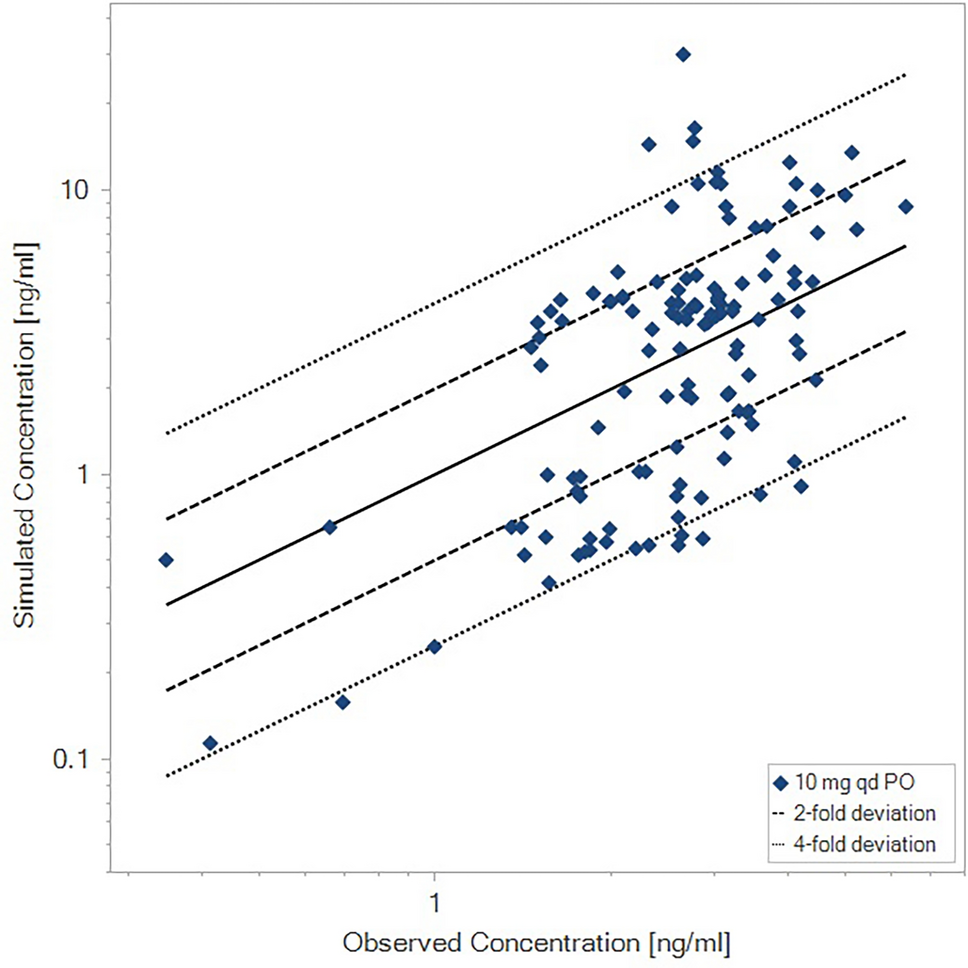
2.9 Population Pharmacokinetic Analysis
A population PK analysis [5] was conducted using pooled data from ulotaront phase I and II studies. The ulotaront population PK dataset was comprised of 404 subjects contributing a total of 4149 plasma ulotaront concentrations. Of the 404 subjects, 99 were healthy volunteers and 305 were patients with schizophrenia. For this analysis population, a two-compartment model with first-order absorption adequately described the data. Ulotaront CL/F values for subjects observed in ulotaront clinical studies were determined by the population PK model.


留言 (0)