
記住我
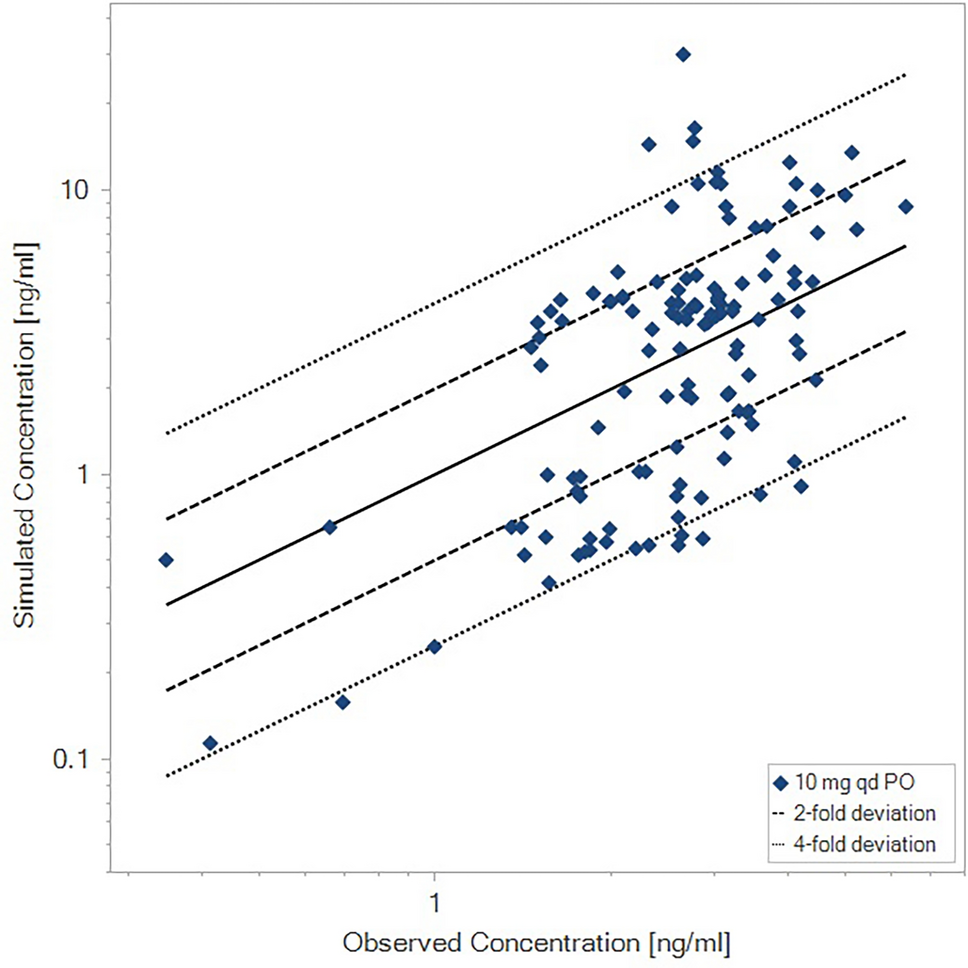

The influence of extrinsic factors is illustrated in Fig. 5 and food effects are discussed in Sect. 3.1, the effect of comedications affecting gastric pH and metabolic DDIs are discussed in Sect. 5.1.1 based on Phase I studies, and in Sect. 5.1.2 based on model-informed approaches.
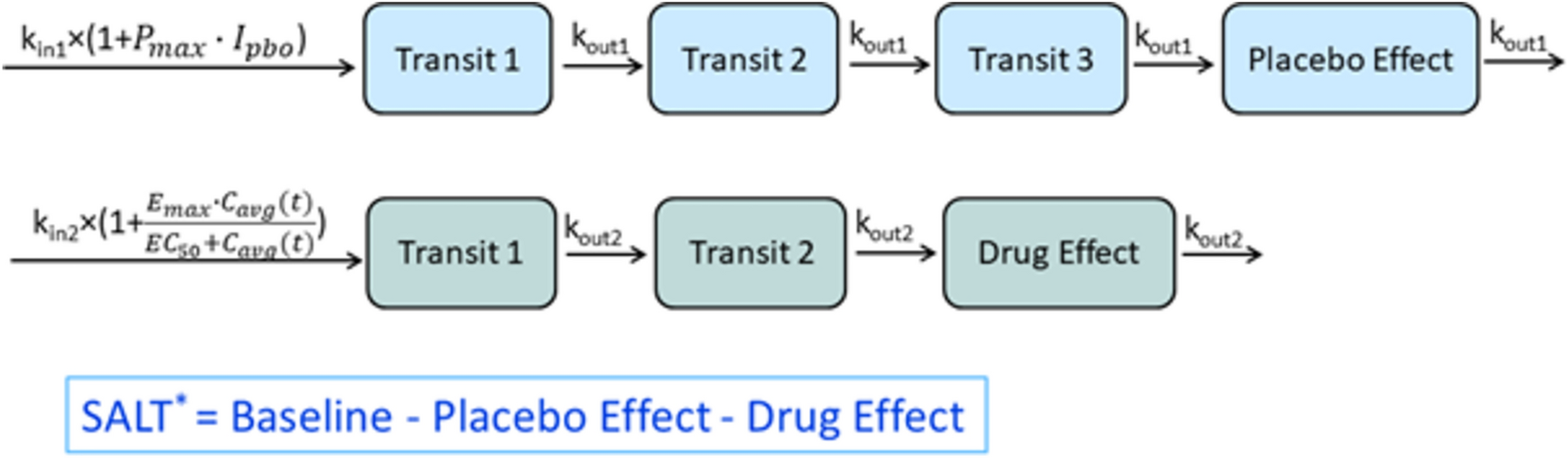
Fig. 5
Effect of extrinsic factors on finerenone exposure; Phase I studies presented as geometric LS mean and 90% CI for ratio “finerenone + extrinsic factor/finerenone alone,” model-informed approaches based on an exploratory Phase IIa popPK covariate effect (amiodarone), and PBPK simulations (other substances) as geometric mean ratios for “finerenone + extrinsic factor/finerenone alone” and range based on sensitivity scenarios assuming ± 5% altered hepatic fm via CYP3A4 and CYP2C8 compared to the reference model with ~90% fm(CYP3A4) versus 10% fm(CYP2C8). AUC area under the curve, BID twice daily, CI confidence interval, Cmax peak drug concentration, CYP cytochrome P450, fm fractions metabolized, LS least squares, OD once daily, TID three times daily, PBPK physiologically based pharmacokinetic, popPK population pharmacokinetic
5.1.1 Phase I StudiesComedications increasing gastric pH were investigated in Phase I studies, as the absorption of finerenone could theoretically be altered given its decreased aqueous solubility with increasing pH. The proton pump inhibitor omeprazole (40 mg on the day of finerenone administration, and 40 mg OD on the previous 4 days) had no effect on finerenone AUC or Cmax. Algeldrate (aluminum hydroxide/magnesium hydroxide 70 mVal suspension 10 mL; Maalox®) did not influence finerenone AUC and decreased Cmax by about 19% [19].
Finerenone was investigated as a substrate/victim in DDIs focusing on CYP3A4 and 2C8 as the only enzymes of relevance for its metabolism. Studies were conducted with index inhibitors [26] of these CYP isoforms, with erythromycin and verapamil as moderate CYP3A4 inhibitors and gemfibrozil as a strong CYP2C8 inhibitor [17]. Erythromycin (500 mg three times a day) reduced finerenone CL/F (−71.3%), resulting in increased AUC (+ 248%), Cmax (+ 88.2%), and t1/2 (from 1.6 to 2.5 h) compared to finerenone alone. Verapamil (240 mg OD) decreased finerenone CL/F (−63.0%), leading to AUC and Cmax increases by 170% and 122%, respectively, and a prolonged t1/2 (from 1.9 to 3.2 h). Changes observed in the concentration versus time profiles of finerenone metabolites M-1, M-2, and M-3 in plasma in the presence of erythromycin or verapamil confirmed the relevance of CYP3A4 for this pathway. Gemfibrozil (600 mg twice daily) reduced finerenone CL/F (−9.1%), resulting in AUC and Cmax increases by 10.1% (90% CI within 80%–125%) and 15.7%, respectively, without effect on t1/2. The effects of erythromycin and verapamil observed in vivo were consistent with results of a static prediction algorithm including the fraction metabolized by CYP3A4 (0.88–0.89 based on in vitro data) and the reported in vivo potencies of CYP3A4 inhibitors [27, 28]. Similarly, the effect of gemfibrozil confirms the fraction metabolized via the CYP2C8 pathway to be approximately 0.10 [17].
5.1.2 Model-Informed ApproachesA PBPK model of finerenone was developed based on physiochemical, in vitro, and clinical Phase I PK data. Informed by preclinical data and clinical Phase I DDI studies, a hepatic fraction metabolized by CYP3A4 of approximately 90% was assumed (reference scenario). As sensitivity scenarios, alternative finerenone models with hepatic fractions metabolized by CYP3A4 constituting 85% or 95% of total hepatic metabolic clearance were implemented as well and used to generate prediction intervals [29].
Model development was conducted in the open-source PBPK platform PK-Sim, qualified for the application of assessing substances as an object of CYP3A4-mediated DDIs [30,31,32,33,34]. The finerenone model was coupled with precipitant/perpetrator models of interest included in the platform without additional adaptions to the models. The coupled models were simulated in 1000 virtual Phase I-like individuals to evaluate the DDI [29].
First, to further validate the finerenone model, especially regarding the contribution of CYP3A4 metabolism to total clearance, DDIs with erythromycin and verapamil were simulated and compared to data observed in dedicated clinical Phase I studies. The simulated versus observed geometric mean ratios for finerenone were 3.46 versus 3.48 (AUCR) and 2.00 versus 1.88 (CmaxR) with erythromycin, and 2.91 versus 2.70 (AUCR) and 1.86 versus 2.22 (CmaxR) with verapamil [29] (corresponding to simulated decreases in CL/F of −71.1% and −65.6%, respectively).
Then, the validated model was coupled to other available precipitant models to predict clinically untested DDIs with various CYP3A4 modulators. An AUCR and CmaxR of 6.31 and 2.37, respectively, were predicted with itraconazole, and of 5.28 and 2.25, respectively, with clarithromycin, in line with the general classification of these drugs as strong inhibitors [35]. This corresponded to simulated CL/F decreases of −84.2% and −81.1%, respectively. An AUCR of 1.59 and a CmaxR of 1.40 was predicted with cimetidine, and of 1.57 and 1.38 with fluvoxamine, corresponding to simulated CL/F decreases of −37.1% and −36.3%, respectively. While the PBPK evaluations with the sensitive CYP3A4 substrate finerenone [36] support a “weak” classification for both cimetidine and fluvoxamine, the latter is labeled “weak to moderate” in Fig. 5 and Table 2, reflecting the FDA’s update of the fluvoxamine classification from a “weak” to a “moderate” inhibitor in 2019. With respect to CYP3A4 induction, an AUCR of 0.19 and a CmaxR of 0.32 were predicted with efavirenz (600 mg OD), and of 0.07 and 0.14 with rifampicin, which are considered moderate and strong inducers, respectively (see Fig. 5 and Table 2) [29]. This would indicate increases in CL/F by > 400% and > 1000%, respectively.
Table 2 Food-drug and drug-drug interactions with finerenone as object of the interaction, summary of extrinsic factorsPredictions pertinent to strong inhibitors and moderate/strong inducers can be considered extrapolations, which are seen as more uncertain than interpolation from stronger to weaker modulation classes. The overall well-qualified models and robust predictions, with the reference scenario (hepatic fm [CYP3A4] = 90%) being closer to observed data than investigated sensitivity scenarios (hepatic fm [CYP3A4] = 85% or 95%), led to the inclusion of the predictions in relevant labels, e.g., US prescribing information [37]. This was complementary to the information based on dedicated Phase I studies and provided a quantitative basis to guide clinical use of finerenone with concomitant CYP3A4 modulators [29].
Apart from PBPK assessments, the use of the weak CYP3A4 inhibitor amiodarone resulted in a 17% lower apparent clearance after forward inclusion corresponding to a 21% AUC increase in an explorative popPK analysis of Phase IIa data [5, 10, 23]. In FIDELIO-DKD, CYP3A4 inhibitor use was identified as a covariate; however, with smaller effects than expected based on the above Phase I and PBPK studies. The discrepancy likely reflects the less controlled and documented study and analysis design as far as the identification of comedication effects in Phase III studies is concerned [24].
Interestingly, in the FIDELIO-DKD popPK analysis, longer-term concomitant use of SGLT-2 inhibitors was identified as a covariate on finerenone CL/F and F, slightly decreasing finerenone exposure. SGLT-2 inhibitor use may improve the general renal pathophysiology in CKD patients and result in decreased levels of uremic toxins and increased CYP3A4 activity [24, 38]. However, additional data are needed to confirm and better understand this incidental finding.
5.2 Finerenone as Precipitant5.2.1 Effect on CYP EnzymesIn vitro studies had indicated a potential for finerenone or some of its metabolites to affect the activity of CYP3A4, both via inhibition (reversible and irreversible) and induction. In addition, a degree of CYP2C8 inhibition was observed in vitro, for which clinical relevance at the exposure level of 20 mg finerenone could not be excluded. Subsequently, studies were performed with the index substrates midazolam (CYP3A4) and repaglinide (CYP2C8) [26]. Although the primary rationale of a DDI study conducted with warfarin was to assess the PDs of this drug with narrow therapeutic range [39], S-warfarin also served as an index substrate of CYP2C9 [26] (see Fig. 6).
Fig. 6
Effect of finerenone on substrate PKs presented as mean and 90% CI for ratio “substrate + finerenone/substrate alone” in dedicated Phase I studies. AUC area under the curve, BCRP breast cancer resistance protein, CI confidence interval, Cmax peak drug concentration, CYP cytochrome P450, h hour, OATP organic anion transporting polypeptide, OD once daily, P-gp permeability glycoprotein, PK pharmacokinetic
Finerenone at the steady-state of its highest approved dose (20 mg OD) had no relevant effect on CYP3A4, i.e., the mean Cmax and AUC of midazolam were increased by 9% and 11%, respectively, and the 90% CI of the AUC ratio was included in the conventional equivalence interval of 80–125% [39]. This observed mean increase in midazolam AUC is comparable to the increase in finerenone AUC following repeated administration (about 10%, see Sect. 3.3), which reflects the common characteristic of midazolam and finerenone being sensitive CYP3A4 substrates. A single dose of 20 mg finerenone administered concomitantly with or 3 h before repaglinide had no relevant effect on repaglinide clearance, considering the demonstrated equivalence in AUC (and Cmax) of the CYP2C8 substrate (90% CIs of repaglinide AUC [and Cmax] ratios within 80%–125%) [39]. In a third study, finerenone (20 mg OD over 6 days) had no effect on AUC and Cmax of R- or S-warfarin, with point estimates and 90% CI of treatment ratios contained within 80%–125%. This confirms the lack of effect of finerenone on CYP2C9, the metabolizing enzyme of S-warfarin. Warfarin also had no effect on the plasma exposure of finerenone, as expected. Finerenone AUC and Cmax ratios (finerenone + warfarin / finerenone point estimates [exploratory 90% CI]) were 0.967 (0.939; 0.997) and 0.933 (0.792; 1.10), respectively [5]. With respect to the PD focus of the study, finerenone did not affect the AUC (0–96 h) of prothrombin time, nor of factor II and factor X activity. For the AUC (0–96 h) of factor VII activity, the upper 90% CI limit of the ratio (1.12) fell above the predefined upper equivalence limit of 1.11, owing to an outlier value in one subject [39].
5.2.2 Effect on Transport ProteinsIn vitro studies have indicated that the permeability glycoprotein (P-gp) and breast cancer resistance protein (BCRP) are inhibited by finerenone [5, 7]. Moreover, finerenone and/or some of its metabolites showed a potential to inhibit the organic anion transporting polypeptide (OATP) isoforms 1B1 and 1B3 [5, 7]. A risk for finerenone to inhibit BCRP and OATP in vivo could not be completely ruled out based on prediction algorithms, while a risk for clinically relevant P-gp inhibition was not evident based on predictive algorithms and predefined threshold values [5]. Studies were performed with finerenone to investigate effects on index substrates of the above drug transporters (see Fig. 6).
Digoxin, an index substrate of P-gp [26], and drug with a narrow therapeutic range, demonstrated equivalence in trough concentration within a dosing interval after multiple dosing (Ctrough,md) on three subsequent days, and in AUC within the dosing interval after multiple dosing (AUCT,md), and Cmax within a dosing interval after multiple dose administration (Cmax,md) (ratios “digoxin + finerenone/digoxin alone” and 90% CIs within 80%–125%), when administered alone or together with finerenone 20 mg OD, with both drugs having reached steady-state. Finerenone AUCτ,md and Cmax,md were also unchanged in the absence or presence of digoxin, as expected [5] (see also Table 3 of the Electronic Supplementary Material).
The AUC or Cmax of rosuvastatin were not altered in a clinically relevant manner when administered on a background of 40 mg finerenone OD at steady-state, simultaneously with finerenone, or separated by 4 h, compared to administration of rosuvastatin alone. Rosuvastatin is an index substrate of both OATPs and BCRP [26]. In addition, finerenone had no effect on the systemic exposure of the endogenous OATP substrates coproporphyrin I and III [7].
The studies show that finerenone does not affect the activity of the drug-transporters P-gp, BCRP, or OATPs 1B1 and 1B3 in vivo.
留言 (0)