
Remember me
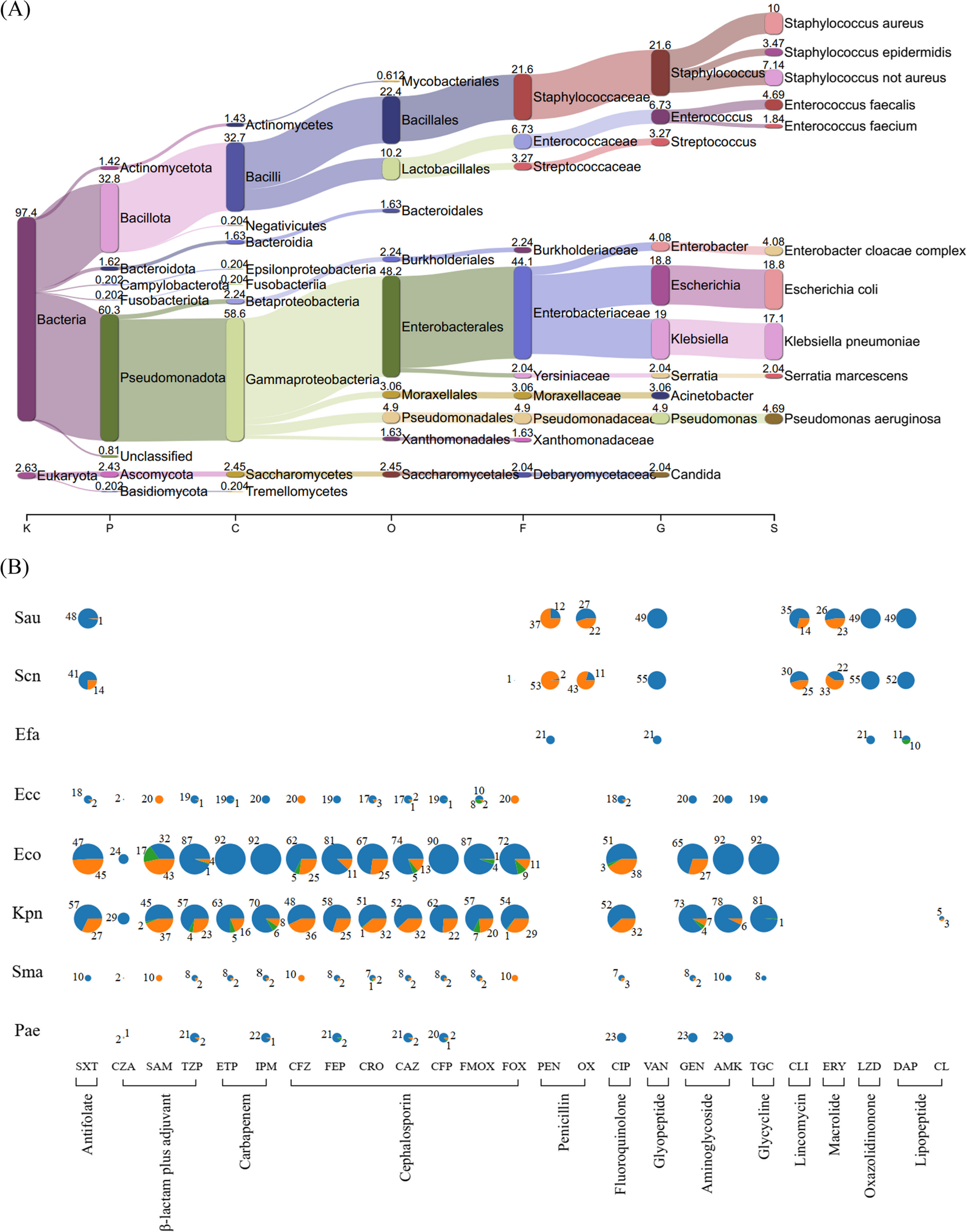



Comprehensive single-cell transcriptome profiling was performed, after thawing viably frozen patient BM samples collected at the time of initial diagnosis (Dx), using the 10x Genomics platform. To robustly map the transcriptome landscape, we also included two MPAL samples from the ScPCA initiative [15]. In total, we performed analysis on nine pediatric MPAL samples (n = 8 at diagnosis, n = 1 at relapse) to understand underlying molecular mechanisms and explore potential therapeutic targets. To identify blast cells and study their transcriptome landscape, the data from healthy pediatric and young adult BM samples was included in the analysis [16]. In total, we analyzed the transcriptome profile of 67,024 cells (23,318 from five T/My MPAL, 17,258 from four B/My MPAL, and 26,448 from healthy BM samples). After quality control, filtering, and normalization, the unsupervised analysis identified 26 transcriptionally distinct clusters of cells (Additional file 2: Fig. S2). The annotation based on the individual samples depicted segregation in the single-cell profiles of MPAL subtypes from healthy BM samples (Fig. 1A). The cellular clusters were labeled based on the expression of canonical cell lineage-associated markers (Fig. 1B); these cell types include erythroblasts, monocytes, T-cells, B-cells, Pro-B, NK, and progenitor cells. The expression of these markers in individual sample’s blast cells is shown in Additional file 2: Fig. S3. The putative MPAL leukemic blast clusters depicted segregated clustering from the healthy normal cell clusters (Fig. 1C, non-blast cells lassoed). B/My MPAL blasts formed segregated clusters as compared to T/My MPAL, indicative of subtype heterogeneity (Fig. 1C). The gene expression of blast markers used in flow cytometry for immunophenotypic characterization is shown in Additional file 2: Fig. S4. On the other hand, immune cells from both MPAL subtypes clustered together with no subtype heterogeneity (Fig. 1C). Further individual sample analysis captured significant heterogeneity among blast cells from some patients (Additional file 2: Fig. S5). For example, T/My MPAL sample M2 and B/My MPAL sample M3 contained two and three distinct blast cell clusters on UMAP visualization respectively, correlating with the characterization on flow cytometry (Additional file 1: Table S2, and Fig. 1D). For the T/My MPAL M2 sample, there were two blast cell populations, which overexpressed myeloid (e.g., S100A8, S100A9, and LYZ) and T-lymphoid (e.g., CD3D and CD3E) lineage markers respectively (Additional file 1: Table S6A). Similarly, DEGs analysis between the two major blast populations in the B/My MPAL M3 sample (Additional file 1: Table S6B) showed over-expression of myeloid (e.g., S100A8, S100A9, and LYZ), and the B-lymphoid (e.g., CD79B, CD79A, and MZB1) lineage markers respectively. These results clearly depict the usefulness of single-cell profiling in highlighting the heterogeneity of blast cell populations, which is a unique attribute of MPAL cases. As anticipated, the profile of cancerous samples collected at the time of initial diagnosis or relapse was primarily composed of malignant blasts (Fig. 1E), with a limited enrichment of normal stromal and immune cell types. Available immune cell type breakdown from diagnostic flow cytometric characterization (Additional file 1: Table S2) correlated with the clusters seen in single-cell profiling. In contrast, the healthy BM samples consisted of major cell types from lymphoid and myeloid lineages including B-cells, T-cells, and natural killer (NK) cells (Fig. 1E).
The differential expression analysis between B/My or T/My MPAL blasts and healthy cells can provide insight into the molecular mechanisms underlying the development and progression of MPAL subtypes. The differential expression analysis based on fold change and p-value (Wilcoxon rank test, average log2FC > 0.25 and adjusted p-value < 0.05) identified 219 and 194 significant DEGs in B/My and T/My MPAL blast cells respectively. The top 20 genes based on average log2 fold change for MPAL subtypes were selected and plotted on a heatmap (Fig. 1F). B/My MPAL blasts depicted significant overexpression genes such as STMN1 and SOX4, which have been previously associated with other hematological malignancies, such as AML and ALL [39, 40]. T/My MPAL blasts overexpressed genes such as SPINK2 which has been associated with immune infiltration in AML [41], and CD82 which has been identified as a driver of chemoresistance in AML [42]. Interestingly, B/My MPAL blasts up-regulated genes show high expression in healthy B/Pro-B-cells (Fig. 1F) indicating that these genes might play a role in the normal differentiation and maturation of B/Pro-B-cells. While T/My MPAL blasts overexpressed genes were observed to exhibit minimal or no expression in the normal T-cells derived from the young adult BM. We did not investigate the expression of these T/My MPAL blasts genes in precursor T-cells from the thymus, which represents a limitation of our study (Fig. 1F). To understand the dysregulations at Pathways and Gene ontology levels, we performed gene ontology (GO) enrichment analyses on the significantly overexpressed genes for the MPAL subtypes (Additional file 1: Table S7). The top significantly enriched (Benjamini–Hochberg adjusted p-value < 0.05) GO categories for B/My MPAL blasts are associated with activation and regulation of the immune response (Additional file 2: Fig. S6A). On the other hand, for T/My MPAL blasts overexpressed genes depicted significant enrichment in GO categories associated with cell signaling and negative regulation of myeloid and leukocyte differentiation (Additional file 2: Fig. S6B).
To further characterize the GO categories that are commonly dysregulated in T/My and B/My MPAL blast cells compared to healthy progenitor cells, we performed additional DEGs analysis comparing MPAL blast cells (n = 35,515) and healthy progenitor cells (n = 3902). The DEGs (Additional file 1: Table S8A, n = 147) were subsequently used to perform GO enrichment analysis to identify associated biological processes. The MPAL blasts overexpressed genes revealed significant associations with myeloid leukocyte migration, NF-kB transcription factor activity, and immune system processes (Fig. 1G). In addition, cellular communication analysis of B/My and T/My MPAL cells via the expression of ligands and receptors using the CellChat tool identified dysregulated signaling pathways associated with aberrant cellular communications. The macrophage migration inhibitory factor (MIF) signaling pathway was found to be enriched in both MPAL subtypes and was associated with signaling from blasts to immune cells including monocytes and B-cells (Fig. 1H). The gene for the ligand of the MIF signaling pathway (MIF) depicted significant up-regulation in both T/My and B/My MPAL blasts compared to healthy cells (Additional file 1: Table S8B). These results mapping the landscape of MPAL subtypes, highlight the inter- and intra-patient heterogeneity in blast cell profiles and similarities in the immune microenvironment landscape.
Compared to other acute leukemias, the scRNAseq profiles of B/My MPAL show significant overlap with B-ALL and AML, while T/My MPAL shows overlap with AML and T-ALLTo assess the similarities and differences between MPAL and other acute leukemias, we performed comparative analyses among MPAL, AML, B-ALL, T-ALL, and healthy BM single-cell profiles. Single-cell transcriptome data for other leukemias (Additional file 1: Table S9) were obtained from the Pediatric Cancers Single-Cell Atlas [17, 18, 43] initiative of our lab and publicly available studies [16]. After uniform pre-processing, filtering, normalization, and batch correction, the gene expression profiles were visualized using UMAP (Fig. 2A). UMAP analysis showed that B/My MPAL blasts clustered mostly with B-ALL and AML (clusters 0, 4, and 6) due to presence of both myeloid and B-lymphoid features in the B/My MPAL blasts (Fig. 2A, Additional file 2: Fig. S7). On the other hand, T/My MPAL blasts profile overlapped with T-ALL and AML (clusters 0, 1, and 8) due to myeloid and T-lymphoid features in the T/My MPAL blast cells (Fig. 2A, Additional file 2: Fig. S7). Interestingly, immune cells clustered based on the cell lineage irrespective of leukemia type/subtype indicating minimal heterogeneity in the immune landscape; T and NK cells are located mostly in clusters in 3, 7, 13, and 24; B-cells in cluster 9; and monocytes in clusters 2, 5, and 16. To evaluate stemness levels, we also calculated the stemness index for the single-cell clusters based on the expression values of 175 genes in the stemness index signature identified by Palmer et al. (Additional file 1: Table S3) [36]. Higher stemness was observed for blast cells from different acute leukemias as compared to non-blast immune cells. The T/My MPAL blast cells showed the highest level of stemness compared to other blast populations, whereas the B/My MPAL blast cells stemness index was similar to B-ALL and AML blasts (Fig. 2B). A sample-wise DEG analysis was performed, to calculate sample-to-sample distances based on the DEGs. The analysis revealed that blast cells from B/My MPAL and B-ALL exhibit the most similar profiles, whereas blast cells from T/My MPAL are most similar to near ETP/ETP T-ALL samples (Additional file 2: Fig. S8).
Fig. 2
Comparative analysis of mixed phenotype acute leukemia with other acute leukemias. A Split UMAP of leukemic and canonical cell types (n = 156,489 cells), separated based on leukemia type/subtype (i.e., AML, B-ALL, T-ALL, B/My MPAL, and T/My MPAL) and healthy samples. B Density plot showing stemness index distribution of the different blast cells from different acute leukemias including B/My MPAL and T/My MPAL, progenitor cells, and normal immune cells. The stemness index was calculated as the first principal component value of each cell after performing principal component analysis with the expression of the genes in a stem cell signature (Additional file 1: Table S3). C Heatmap with the top overexpressed markers for mixed phenotype acute leukemia (MPAL) and subtypes (i.e., B/My MPAL and T/My MPAL). The heatmap also shows the expression of MPAL marker genes in other acute leukemias (i.e., AML, B-ALL, T-ALL), (BM) and healthy immune cells. These markers were filtered to only include genes with low expression in healthy bone marrow cells. Overexpressed genes were identified for MPAL subtypes by comparing the profile of MPAL blast cells versus blast cells from other acute pediatric leukemias (i.e., AML, B-ALL, T-ALL) and healthy BM samples. The MPAL subtype significantly overexpressed genes (average log2FC > 0.25, adjusted p-value < 0.05, and pct. expressed > 50%) were further refined by selecting genes with low expression in healthy BM cells from HCA (avg. expression < 0.5). Finally, the top genes for the heatmap were chosen based on their highest average log2FC values. D Dot plots showing the expression of two canonical immune cell markers (CD79A and CD3D) and two MPAL blast cell markers (CD81 and LMO2), to show that these MPAL blast cells markers have low expression in various normal BM cell types and healthy hematopoietic stem cells. The size of the dots refers to the percentage of cells in each cell type cluster expressing the gene and the color represents averaged scaled gene expression level; cyan: low, red: high. X-axis is the cell type, and Y-axis is the genes. The expression of MPAL markers is marked with lasso. E Expression of MPAL blast markers in AML, T-ALL, and MPAL bulk RNA-seq data. The Y-axis shows the scaled values of the log2 of the normalized expression plus one, and the X-axis shows different subtypes for the bulk RNA-seq samples. Wilcoxon rank tests were performed to test the difference in expression between MPAL and AML, and MPAL and T-ALL for the three genes shown (*** for p-value < 0.001, ** for p-value < 0.01, and * for p-value < 0.05). F The top significantly enriched pathways of the filtered B/My MPAL blast cell marker genes. Each bar represents a significantly enriched pathway as determined using the P value (shown on the primary X-axis). The bar plot is sorted by the negative log of the hypergeometric distribution-based p-values of the results. The analysis for canonical pathways was performed using the MetaCore platform from Clarivate Inc. G The top significantly enriched pathways of the filtered T/My MPAL blast cells marker genes. H Kaplan–Meier curves-based survival association analysis of B/My MPAL marker gene, MTRNR2L12 in B/My MPAL TARGET samples (top) and T/My MPAL marker, PTEN in T/My MPAL TARGET samples (bottom). Survival association analysis was performed using the Cox Proportional Hazards Regression Model, with MTRNR2L12 expression having a hazard ratio of 4.80 (p = 0.059) and PTEN expression having a hazard ratio of 4.50 (p = 0.04), high expression of both genes indicated an association with poor survival
To identify genes with significant overexpression in MPAL as compared to other leukemias and healthy controls, DEG analysis was performed based on the Wilcoxon rank test (FC > 0.25 and adjusted p-value < 0.05). Genes that are overexpressed in both MPAL subtypes blast cells compared to other leukemias and healthy controls include CD81, and LMO2 (Fig. 2C). B/My MPAL blast cells overexpressed genes include AUTS2, PAX5, MTRNR2L12, and HBEGF. T/My MPAL blast cells overexpressed genes include PTTG1IP, ESYT2, PTEN, and CALCOCO2. To ensure markers are MPAL blast cell-specific, we performed additional filtering steps based on expression in the BM immune cells and hematopoietic stem cells (HSCs). The BM single-cell data of 391,505 immune cells and HSCs were obtained from the Human Cell Atlas (HCA) Initiative [44]. MPAL blast-specific genes were found to be minimally expressed in these HSCs and immune cells from HCA (Fig. 2D) in contrast to canonical immune cell markers such as CD3D for T-cells and CD44 for HSCs, which had high expression in these cells. Interestingly, CD81 had relatively high expression in the BM mesenchymal stem cells (MSCs) that constitute 0.06% (n = 239 cells) of the HCA dataset (Fig. 2D). Recent studies have shown compelling evidence that CD81 serves as a promising marker for identifying MSC-derived extracellular vehicles and intracellular communication [45]. The complete lists of MPAL blast markers with fold change and p-values from each step of the filtering process (Additional file 2: Fig. S1) are listed in Additional file 1: Table S4. The biomarker genes, CD81 and LMO2 which are expressed in both MPAL subtypes (Fig. 2D) had minimal or no expression in normal immune and stem cells, making these genes ideal potential candidates for targeting. In addition, the expression of the B/My and T/My MPAL biomarker genes was also assessed (Additional file 2: Fig. S9A, B) using the TARGET bulk RNA-seq MPAL, AML, and T-ALL datasets [31]. Some of the MPAL subtype blast markers showed higher expression in MPAL bulk RNA-seq data, and the rest showed noisier results. We do not expect the bulk-RNA-seq data to match our single-cell expression patterns completely as bulk RNA-seq data depicts the average expression profile of immune, stromal, and blast cells in a sample. The selected genes (CHST11, GLS, PPP1R12A) that are overexpressed in B/My and T/My MPAL blast cells in single-cell data, showed significantly higher expression (Wilcoxon rank test, p-value < 0.05) in MPAL versus T-ALL and AML bulk RNA-seq TARGET samples (Fig. 2E).
To further understand the potential pathway level dysregulation in MPAL blast genes, pathway enrichment analysis was performed on the MPAL subtypes (i.e., T/My, B/My MPAL) blasts significantly overexpressed genes (Additional file 1: Table S4). The pathways analysis depicted the significant enrichment (p-value < 0.05) of B/My MPAL overexpressed genes in multiple cell cycle, proliferation, and immune system-related pathways (Fig. 2F). This included activation of protein kinase C (PKC) via G-Protein coupled receptor, gastrin signaling in inflammatory response, angiotensin II receptor type 1 (AGTR1) signaling via p38, extracellular-signal-regulated kinase (ERK) and epidermal growth factor receptor (EGFR) signaling, and glucocorticoid receptor signaling, as well the role of IL8 typically seen in colorectal cancer (Fig. 2F). Similar analysis on T/My MPAL overexpressed genes depicted a significant association with cell cycle, cell adhesion, and immune response including tissue factor signaling, ERK1/2 signaling, IL6 signaling similar to that seen in prostate cancer, and PTEN pathways (Fig. 2G). The pathway enrichment analysis also depicted a significant (p-value < 0.05) effect on Sphingosine 1-phosphate receptor 2 (S1P2) signaling in both B/My and T/My MPAL (Fig. 2F, G). Interestingly, while the activation signaling of S1P2 was prominent in B/My MPAL, the inhibitory signaling was most enriched in T/My MPAL. This dissimilarity in S1P2 signaling could be attributed to the upregulation of HBEGF in B/My MPAL and PTEN in T/My MPAL (Fig. 2D). HBEGF is a downstream target of S1P2 signaling, resulting in the promotion of cell survival (Additional file 2: Fig. S10A); whereas PTEN is a direct target of S1P2 signaling, resulting in the inhibition of FAK1 and inhibition of cell migration (Additional file 2: Fig. S10B). The analysis on top B/My MPAL affected pathways depicted that HBEGF was involved in 10 out of 15 top enriched pathways (Fig. 2F), whereas PTEN was found to be involved in 12 out of the 15 top affected pathways in T/My MPAL (Fig. 2G). The complete lists of significantly enriched pathway maps along with p-values are listed in Additional file 1: Table S10. To further explore the role of MPAL blast cells overexpressed genes in cancer outcomes, we performed survival analysis using the Cox proportional hazards model in the TARGET-ALL-P3 dataset [31]. The high expression of the B/My MPAL blast marker MTRNR212 had an association with poor survival (HR = 4.80, p-value = 0.059) in B/My MPAL samples. Whereas high expression of PTEN, a T/My MPAL blast marker gene depicted a significant association with poor survival (HR = 4.50, p = 0.040) in the T/My MPAL samples (Fig. 2H).
T/My MPAL has higher similarity to ETP than with non-ETP T-ALL, but still displays unique myeloid characteristicsGiven the emerging literature showing greater overlap between T/My MPAL and ETP-ALL, we decided to compare the single-cell transcriptomic profiles between the two groups as well as non-ETP T-ALL cases. Among the 11 T-ALL cases used in the analysis, four cases had ETP-ALL-like features (ETP-ALL, n = 1 and near ETP-ALL, n = 3), while the remaining seven were categorized as non-ETP T-ALL (Additional file 1: Table S11). Among the five T/My MPAL cases, three had ETP-like immunophenotypic blasts features on flow cytometry, aside from their myeloid lineage defining antigen expression, that resulted in a diagnosis of T/My MPAL (Additional file 1: Table S2).
To assess the scRNAseq similarities of T/My MPAL to T-ALL and ETP subtypes, we performed a focused analysis on these 16 samples (Fig. 3). The UMAP visualization depicted T/My MPAL, ETP and T-ALL formed distinct clusters with some overlaps among them (Fig. 3A). For the blast populations specifically, there exist clusters with cells from T/My MPAL and ETP/near-ETP (cluster 0) and T/My MPAL and non-ETP T-ALL (cluster 7), as well as a distinct myeloid cluster 3 (high expression of S100A9, LYZ, CD14) which contains mostly T/My MPAL blasts (Fig. 3B). Differential expression analysis was performed to determine the top overexpressed genes for near-ETP/ETP-ALL blast cells compared to non-ETP T-ALL and T/My MPAL blast cells compared to non-ETP T-ALL. This analysis found that there are 353 common genes overexpressed in near-ETP/ETP-ALL and T/My MPAL compared to T-ALL (Fig. 3C). An additional analysis was performed to compare the three groups, with AES and CD3D showing high expression in non-ETP T-ALL blast cells, and near-ETP/ETP blast cells over-expressing CKLF and TASP1 (Fig. 3D). T/My MPAL blasts overexpress VAMP8 and SAT1 with high specificity compared to the other T-ALL blast cells (Fig. 3D). The DEG analysis identified 1,021 T/My MPAL, 639 near-ETP/ETP, and 831 non-ETP T-ALL overexpressed genes for blast cells (average log2FC > 0.25 and adjusted p-value < 0.05). GSEA was performed on these three sets of genes to determine which biological processes were significantly over-represented (adjusted p-value < 0.05) in each of the three subtypes’ blast cells. GSEA analysis identified T/My MPAL overexpressed gene enrichment in inflammatory and cell growth-related processes (Additional file 2: Fig. S11A). The non-ETP T-ALL blasts had high enrichment of cell differentiation processes (Additional file 2: Fig. S11B), and near-ETP/ETP overexpressed genes linked to transcription regulation and/or structural organization processes (Additional file 2: Fig. S11C) such as PTEN, TNIK, and AUTS2 (Fig. 3E). Further stemness analysis interestingly revealed that near-ETP/ETP T-ALL blasts had the highest stemness index, followed by T/My MPAL and non-ETP T-ALL blasts. The T/My MPAL depict bimodal distribution of the stemness index ranging from high (similar to ETP) to low stemness (similar to T-ALL) indicating heterogeneity at the stemness level (Fig. 3F). To our knowledge, this is the first comparison of T/My MPAL and ETP-ALL on a single-cell transcriptomics level.
Fig. 3
Mapping the single-cell landscape of early T-cell precursor acute lymphoblastic leukemia (ETP-ALL). A UMAP clusters of 50,907 cells colored based samples and different ALL (left) including T/myeloid mixed phenotype acute leukemia (T/My MPAL), near-ETP/ETP-ALL, and non-ETP T-ALL. The right side is the UMAP colored by clusters obtained based on K-mean clustering using the Seurat package. B Cell type annotations for the three T-Lineage subtypes shown on UMAPs. Clusters with the overlap of cells and transcriptome profiles among different T-ALL subtypes have been lassoed and labeled. C Venn diagram analysis to visualize commonly overexpressed genes (average log2FC > 0.25, adjusted p-value < 0.05) in T/My MPAL compared to non-ETP T-ALL blast cells, and near-ETP/ETP-ALL compared to non-ETP T-ALL blast cells. D Feature map of selected T/My MPAL, non-ETP T-ALL, and near-ETP/ETP-ALL blast cells overexpressed genes. Low and high expressions are shown with gray and purple colors respectively. E Gene network plot for enriched GO categories associated with overexpressed near-ETP/ETP-ALL genes. The network nodes have been colored based on fold change in near-ETP/ETP-ALL, and the size of the central dots represents the size of the selected GO category. F Density plot showing stemness index distribution of blast cells T/My MPAL, near-ETP/ETP-ALL, non-ETP T-ALL, and non-blast immune cells. The stemness index was calculated as the first principal component value of each cell after performing principal component analysis with the expression of the genes in a stem cell gene set as the features (Additional file 1: Table S3)
Adult and pediatric MPAL have similar transcriptional landscapesTo determine the similarities and differences between adult and pediatric MPAL, we performed a comparative analysis by analyzing adult MPAL scRNAseq data (n = 6) from a publicly available study [14] (Additional file 1: Table S9). Adult MPAL data after normalization and quality control analysis was merged with pediatric MPAL samples and healthy BM. After performing clustering, annotation, and UMAP visualization inspection, we observed that the adult and pediatric samples predominantly clustered based on the MPAL subtypes rather than by age. This suggests that the MPAL subtypes are a more significant factor in determining the gene expression patterns than the age of the patients (Fig. 4A). In the UMAP, we observed that pediatric B/My MPAL samples (M1, M3, M5, M7) cluster together with the adult B/My MPAL sample (A4) in the lower left part of the plot. Specifically, these samples were distributed in clusters 2, 5, 9, 10, and 18. Similarly, the pediatric T/My MPAL samples (M2, M4, M6, SCPCS000230, SCPCS000220) cluster with the adult T/My MPAL samples (A1, A2, A3, A5, A5R) in the upper right portion of the UMAP in clusters 0, 1, 6, 8, and 27. This finding suggests a possible commonality in gene expression patterns between pediatric and adult MPAL subtypes. Furthermore, immune cells from adult and pediatric samples cluster together and are segregated based on cell type irrespective of age and MPAL subtypes (Fig. 4B).
Fig. 4
Comparative analysis of pediatric and adult mixed phenotype acute leukemia single-cell landscape. A Split UMAP plots of B/My MPAL and T/My MPAL colored based on the respective patient samples. The adult MPAL samples are represented in shades of blue and green, while the pediatric are depicted in shades of red and pink. B Comparative visualization of malignant blasts and normal microenvironment cell types in the adult, pediatric, and healthy samples. C Heatmap of top genes overexpressed in adult vs. pediatric MPAL blast cells. Genes were identified by performing differential expression analysis selecting genes with average log2FC > 0.25 and adjusted p-value < 0.05. The top genes for the heatmap were selected based on average log2FC. Relative gene expression is shown in pseudo color, where purple represents downregulation, and yellow represents upregulation. D Density plot showing the distribution of stemness index of different adult and pediatric MPAL subtypes and normal cells. Density plot showing stemness index distribution of the different cell types found in T/My MPAL samples. The stemness index was calculated as the first principal component value of each cell after performing principal component analysis with the expression of the genes in a stem cell gene set as the features (Additional file 1: Table S3). E Selected gene sets with significantly higher enrichment (p-value < 0.001) in adult T/My MPAL blast cells. F Gene sets with higher enrichment (p-value < 0.001) in pediatric versus adult T/My MPAL blast cells. The enrichment score was calculated using a single-sample gene set enrichment approach using Hallmark/Biocarta gene sets from the MSigDb H and C2 collections and the significance of differential enrichment was determined using the Wilcoxon rank-sum test
To identify subtle potential age-specific MPAL blast markers, we performed differential expression analysis between adult and pediatric MPAL blasts (average log2FC > 0.25, adjusted p-value < 0.05). Heatmap with the top 20 markers (by average log2FC and percent expression) is shown in Fig. 4C. These genes might be linked to age instead of the malignant nature of the blasts. To further assess of stemness of blast cells from different pediatric and adult MPAL subtypes, we performed stemness analysis using a stem cell gene signature from literature [36]. In both MPAL subtypes, the adult patient blasts had higher stemness index values (Fig. 4D), indicating that pediatric MPAL blasts are more differentiated than adult MPAL blasts. Gene set enrichment analysis on pediatric and adult T/My MPAL blast cells provided insights into underlying biological mechanisms. Adult blast cells are significantly enriched (Wilcoxon rank-sum test, p-value < 0.001) with the inflammatory response and the KRAS up-regulated signaling pathway (Fig. 4E), while pediatric T/My MPAL blast cells are significantly enriched (p-value < 0.001) with TGF-β and PTEN pathways (Fig. 4F).
Overall, a comparison of adult and pediatric MPAL subtypes revealed a similar transcriptional landscape with subtle immune and inflammatory pathway level differences that might be due to the high mutational burden in adult MPAL and poorer outcomes.
Diagnostic MPAL samples have transcriptomic differences that may help predict response to ALL induction therapyGiven the growing consensus that MPAL patients should be initially treated with an ALL-directed induction regimen, we explored the association between transcriptome profiles at baseline and the end of induction (EOI) results following an ALL-based chemotherapy. Of the eight de novo MPAL cases, seven received initially an ALL-based induction regimen (Additional file 1: Table S1). These cases were partitioned into different groups based on their outcome status at the end of induction (i.e., MRD + , MRD − , and induction failure) (Table 1). Among the two MPAL induction failure patients, one had a poor initial response to ALL induction and was switched to AML therapy at day 13 (M7), hence was classified as an induction failure to ALL therapy. Given the distinct transcriptome profiles between the B/My and T/My MPAL subtypes, we chose to analyze the two groups separately. Among the B/My MPAL patients, two were MRD + (M3 and M5), one was MRD − (M1), and one had an induction failure, requiring change in therapy (M7). For the T/My MPAL patients, one had induction failure (M6), one was MRD + (M4), and one was MRD − (M2). As this analysis exclusively incorporates samples obtained at the time of diagnosis, the term “blasts from MRD + patients” refers to blast cells identified during the initial diagnosis of patients who were determined to have minimal residual disease (MRD +) at the end of induction (EOI). Clustering and UMAP embeddings based on induction results formed patient-specific clusters in B/My MPAL as well as T/My MPAL (Fig. 5A). One of the B/My MPAL MRD + (M5) patients formed a distant cluster indicating the highest transcriptional difference as compared to the cells from other MRD + , MRD − and induction failure patients. Additionally, MRD + patients also depicted significant heterogeneity in the blast cell profile as evident from multiple blast clusters of the same patient (Fig. 5B). The non-blast cells formed mostly overlapping clusters except monocytes (Fig. 5B). To identify baseline differences in the blasts based on induction status (MRD + , MRD − , induction failure), we performed differential expression analysis for B/My and T/My MPAL samples (Fig. 5C). B/My MPAL blast cells from induction failure patients showed high expression of MT2A and FKBP5 genes that are associated with chemoresistance in osteosarcoma [46], solid cancers, and ALL [47]. The blast cells from the MRD − patients depicted the highest expression of IGHM, a gene associated with good prognosis in breast cancer [48], and IGFBP7, a marker of leukemia cell and chemosensitivity in AML [49]. Blast cells from B/My MPAL MRD + patients overexpressed genes such as NEAT1 and SOX4 that are associated with cancer development and pan-cancer poor outcome [50, 51]. For T/My MPAL the top marker genes for blast cells from induction failure and MRD + patients show significant overlap, whereas markers for blast cells from MRD − patients are more uniquely expressed. To further identify the key pathways over-represented in the three blast groups, we performed gene set enrichment analysis using canonical pathways gene sets from the MSigDB database [52]. Blast cells of B/My MPAL the induction failure patient depicted the highest enrichment (Wilcoxon ranked test, p-value < 0.001) of MAP3K8/TPL2 dependent MAPK1/3 activation (Fig. 5D). B/My MPAL blast cells from the MRD − patient had high enrichment of the translation factors gene set (Wilcoxon ranked test, p-value < 0.001) (Fig. 5D), whereas MRD + blast cells had high enrichment of the PI3K/AKT/mTOR VITD3 signaling pathway (Fig. 5D). Blast cells from T/My MPAL MRD − patient depicted significantly higher enrichment of the cell differentiation expanded index as compared to blast cells from MRD + and induction failure patients (Wilcoxon ranked test, p-value < 0.001) (Fig. 5E). Induction failure and MRD + patient blast cells showed higher enrichment of Stathmin pathway as compared to blast cells from MRD − patients (Fig. 5E). The comparative analysis of stemness index revealed a broader distribution of stemness in T/My MPAL blast cells T/My MPAL compared to ALL and AML patients, indicat
Comments (0)