
記住我





Functional somatic syndromes (FSS) are characterized by medically unexplained symptoms, which cause significant distress and impairment (1,2). Among the most prominent FSS are chronic fatigue syndrome (3), characterized by fatigue and flu-like symptoms; fibromyalgia syndrome (4), characterized by widespread pain; and irritable bowel syndrome, characterized by abdominal pain and digestive problems (5). More than 22% of individuals from the general population report at least one medically unexplained symptom causing severe impairment (6). In primary care, medically unexplained symptoms make up 36% of all health care visits (7). This proportion is even higher in secondary care, such that an unequivocal medical explanation cannot be established in more than 52% of new admissions (8). Unfortunately, these numbers are likely to increase in the aftermath of the COVID-19 pandemic (e.g., (9)). Indeed, critical life events, including viral and bacterial infections, represent one of the most frequent precipitating factors in these conditions (e.g., (10)). Therefore, it is crucial to gain a deeper understanding of the pathophysiological processes that underlie these debilitating illnesses.
The pathophysiology of FSS is multifactorial and has spurred extensive research in the past decades (11). Stress (e.g., childhood trauma, chronic stress, critical life events) and alterations in stress-responsive bodily systems represent significant factors in the development and maintenance of FSS (e.g., (12–14)). The most frequently studied stress-responsive bodily system is FSS, likely due to its pivotal role in regulating processes that can contribute to the experience of medically unexplained symptoms (15). For instance, states of diminished arousal and diminished activity in descending pain inhibitory pathways at the central level can develop by means of interactions of the hypothalamic-pituitary-adrenal axis with the central locus coeruleus/noradrenaline system (16,17). Moreover, in the periphery, failure of the hypothalamic-pituitary-adrenal axis end product, the glucocorticoid cortisol, to exert its gluconeogenetic and anti-inflammatory effects (see Refs. (18–20) for meta-analyses on chronic low-grade inflammation in FSS) impacts energy metabolism and induces a sickness behavior-like state characterized by fatigue, sleep disturbances, and hyperalgesia (16,21).
Building on these findings, epigenetic phenomena, which represent relatively stable biological alterations that can manifest as a result of environmental factors, including stress, have increasingly attracted the attention of researchers in FSS. The best-understood epigenetic phenomenon is DNA methylation, which can be reliably measured in various tissues, including whole blood, peripheral mononuclear cells (PBMCs), and specific cell types, such as T cells (22,23). In brief, DNA methylation refers to the process of methyl molecules attaching to the 5′ carbon position of cytosine residues, which are often present in the promoter region of genes and have functional implications regarding gene expression. In these regions, high levels of methylation are mostly associated with reduced gene expression. However, methylation in other gene regions, such as exons or introns, can also be associated with less pronounced gene expression. As extreme and/or persistent stress can induce chemical modifications of DNA methylation, the question arises of whether DNA methylation may be systematically altered in conditions in which stress is involved (such as FSS), and contribute to symptoms such as fatigue and pain via altered gene expression.
The aim of this study was to provide the first systematic review of the emerging literature on DNA methylation signatures of FSS. We expected that individuals with FSS would be characterized by differential DNA methylation in genes encoding for biological systems previously associated with FSS (e.g., (12–14)).
METHODSMOOSE guidelines (24) were followed, and the study was preregistered on PROSPERO (CRD42022364720). This is a systematic review, and all data were obtained from published articles cited in the article.
Search and Selection of StudiesMEDLINE and PsycINFO were systematically searched from the first available date to September 2022. The search was composed of a) the term “functional somatic syndrome,” as well as synonyms and related terms (i.e., “chronic fatigue syndrome,” myalgic encephalomyelitis,” “fibromyalgia,” “irritable bowel syndrome,” “colon irritabile”), and b) “DNA methylation.” The search was complemented by manual searches of reference lists of all included full texts as well as by a search of Gene Expression Omnibus for published data sets and additional studies of potential relevance.
Studies meeting the following eligibility criteria were included in our systematic review: a) included adults fulfilling the Oxford, Fukuda, or Canadian criteria for chronic fatigue syndrome (3,25,26), American College of Rheumatology criteria for fibromyalgia syndrome (4,27,28), and/or Rome criteria for irritable bowel syndrome (5,29); b) included a healthy control group; and c) included a candidate-gene or genome-wide study of DNA methylation. The former approach targets a specific gene or several genes that encode proteins presumably involved in the pathophysiology of FSS, whereas the latter approach is unbiased and usually covers more than 450,000 or more than 850,0000 sites within the genome. Only research published in English, German, Dutch, Italian, Spanish, Portuguese, or French was considered. Individuals with comorbid illnesses and/or medicated individuals were included, but the FSS had to be the primary disorder, and comorbidity and medication intake were recorded for risk of bias assessments. All tissues (e.g., whole blood, T cells) used for DNA methylation analyses were included. All duplicates were discarded, and the titles and abstracts of all search hits were screened before full-text articles of potentially relevant studies were screened. Although the preregistration mentions that two study investigators would perform the screening, we realized after the initial screening that the straightforward search and small body of literature, which was extremely rich in cross-references, made this redundant.
Data Extraction and Risk of Bias AssessmentFor each included study, information about the first author, the year of publication, the number of patients and healthy controls, eligibility criteria, DNA methylation assessment, and results corrected for multiple testing were extracted. In the case of missing summary statistical data, the authors of the respective study were contacted. This happened once, but no additional data were retrieved as a result of this contact. Risk of bias was assessed using a modified quality assessment scale that was used in previous meta-analyses on stress-responsive systems in FSS and on polymorphisms in genes related to the HPA axis as predictors of antidepressant treatment response (30,31) (Supplemental Digital Content 1, https://links.lww.com/PSYMED/A956). The instrument contained five items, each rated on a scale from 0 to 2. The items covered characteristics of the FSS and healthy control samples (items 1 and 2), the adequacy of DNA methylation methodology and data processing (item 3), the blinding of lab personnel (item 4), and the consideration of important statistical confounders (item 5). The maximum attainable score was 10. Data extraction and risk-of-bias assessment was conducted by two study investigators, and any conflicts were resolved with a third study investigator. κ was 0.83.
Data AnalysisStandardized mean differences were computed based on means and standard deviations of β values, which are used to indicate differences in DNA methylation at specific sites of the genome. Only studies using the same methodological approach (i.e., candidate-gene or genome-wide) and studies using the same sampling tissue (e.g., whole blood) were integrated (the latter criterion was not preregistered, but was necessary because of the cell-type specificity of DNA methylation). Hedges g and 95% confidence intervals (CIs) were to be calculated, and the studies were to be weighed and integrated based on their inverse variance. Random-effects meta-analyses were to be computed using SPSS 28 and metafor (R Core Team, 2022). Heterogeneity was to be estimated using I2 and χ22 statistics. Subgroup analyses based on the items of the quality assessment scale were to be undertaken to study potential effect modifiers. Funnel plots, Egger regression test, and a trim-and-fill procedure were to be used to examine publication bias in the case of more than 10 available studies.
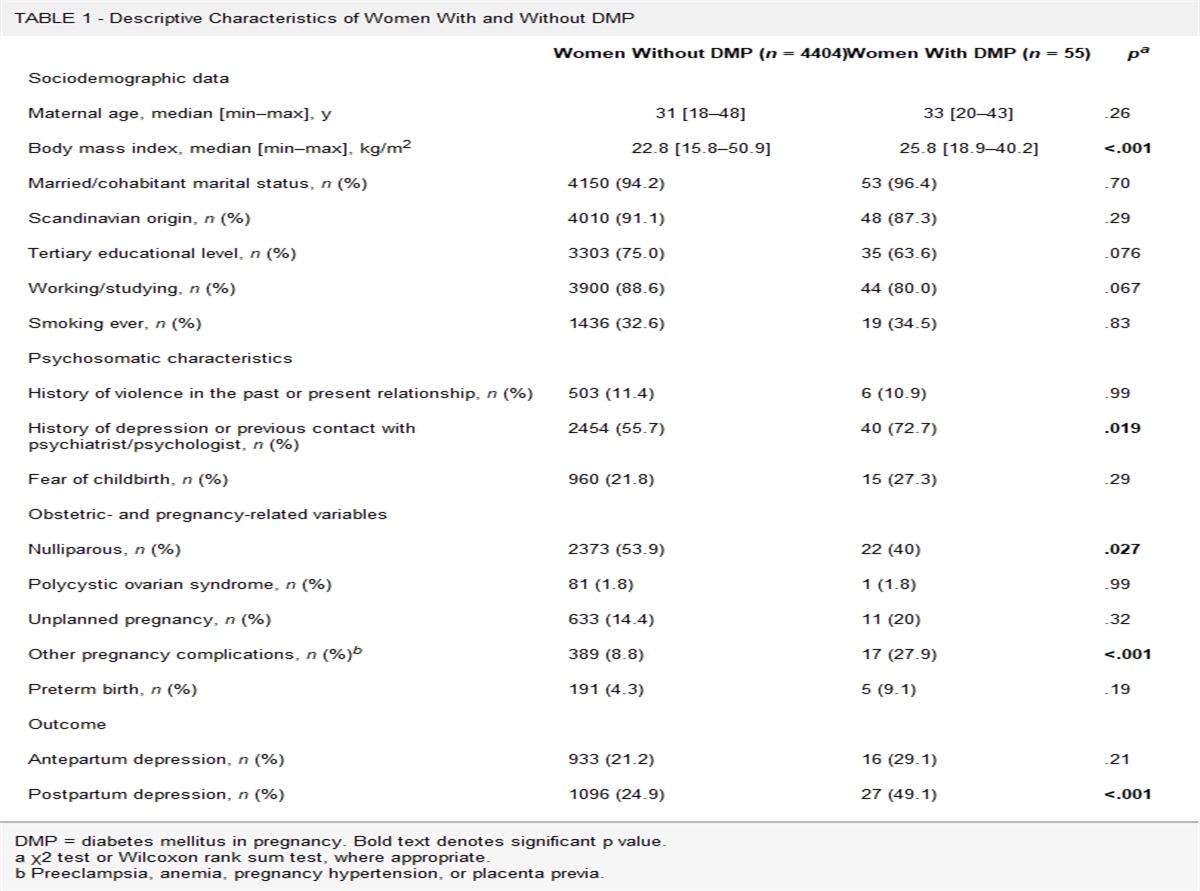
RESULTS Study CharacteristicsThe search process is depicted in Figure 1. The literature search yielded 835 hits, of which 817 were considered irrelevant after screening of the titles and abstracts. A total of 19 full texts were scrutinized for eligibility, and 3 were excluded for the following reasons: no FSS according to the required research diagnostic criteria or no DNA methylation assessment. In total, 16 studies (N = 957) were included in the systematic review.
 FIGURE 1:
FIGURE 1: PRISMA flowchart of study selection.
The characteristics of the studies are summarized in Table 1. In brief, the included studies were published between 2011 and 2021. Most (around two-thirds) of the studies were conducted in chronic fatigue syndrome, followed by fibromyalgia syndrome and irritable bowel syndrome. To diagnose chronic fatigue syndrome, all but one study, which used the Canadian criteria (25), used the Fukuda criteria (3). Moreover, in four of the studies using the Fukuda criteria, the participants also fulfilled the Canadian criteria. To diagnose fibromyalgia syndrome, all but one study, which used the 1990 American College of Rheumatology criteria (27), used the 2010 American College of Rheumatology criteria (4). To diagnose irritable bowel syndrome, Rome III criteria were used (5). Seven studies used a candidate-gene approach, and 9 studies used a genome-wide approach. The average quality assessment score of the studies was 5 of 10, reflecting low to moderate risk of bias. The description of DNA methylation analysis and the consideration of confounding variables, including age, sex, and body mass index, were generally excellent. However, some of the studies did not include smoking as a confounder and did not provide sufficient information on whether individuals with FSS had comorbid somatic diseases or mental disorders or were on medication. Moreover, information on the blinding of the staff involved in DNA methylation analyses regarding participant group status was generally lacking.
TABLE 1 - Candidate-Gene and Genome-Wide Studies Comparing DNA Methylation Between Cases With FSS and Controls Study Cases With FSS Controls DNA Methylation Assessment Results Quality Rating (0–10) Falkenberg et al. (32) n = 49FSS = functional somatic syndrome; PBMC = peripheral blood mononuclear cells; HTR2A = 5-hydroxytryptamine receptor 2a; PRF1 = perforin 1; NR3C1 = nuclear receptor subfamily 3 group C member 1; SNCAIP = synuclein alpha interacting protein; BDNF = brain-derived neurotrophic factor; GCSAML = germinal center associated signaling and motility like.
Seven studies used a candidate-gene approach to examine DNA methylation in FSS. Of these, five used whole blood as a tissue and two used PBMCs.
Chronic Fatigue SyndromeTwo studies in women with chronic fatigue syndrome investigated whole blood methylation of NR3C1, the gene encoding the glucocorticoid receptor, and specifically the exon 1F promoter region (37,43). Of these, the study investigating seven sites showed lower methylation levels regarding one site (denoted “CpG1.5”) at the end of the exon 1F promoter in cases as compared with controls (large effect) (37). The other study, investigating 18 sites (which did not include all of the seven previously investigated sites) found comparably lower methylation levels of an additional site (denoted “CpG8”) located at the end of the exon 1F promoter (large effect) (43). Meta-analysis of all sites overlapping did not reveal any group differences: CpG12.13 (n = 264, g = −0.12, 95% CI = −0.56 to 0.32; z = −0.54, p = .590), CpG17.18 (n = 254, g = −0.17, 95% CI = −0.44 to 0.10; z = −1.26, p = .210), CpG19 (n = 265, g = 0.12, 95% CI = −0.70 to 0.95; z = 0.29, p = .771), CpG38.39 (n = 252, g = 0.10, 95% CI = −0.17 to 0.36; z = 0.71, p = .480), or CpG47 (n = 266, g = −0.09, 95% CI = −0.47 to 0.30; z = −0.44, p = .662). One study in women with both chronic fatigue syndrome and fibromyalgia syndrome investigated whole blood methylation of 44 sites within BDNF, encoding the brain-derived neurotrophic factor, and specifically its promoter 1, exon 3, promoter 4, and exon 9 regions (45). This study found lower methylation of five sites within the exon 9b in cases compared with controls.
One study in chronic fatigue syndrome found no differences in PBMC methylation of 17 sites located within the promoter of HTR2A, encoding the serotonin 5-HT2A receptor, in comparison with controls (32). Another study examined seven sites within the promoter of PRF1, the gene encoding perforin 1, and did not find any differences in PBMC methylation in comparison to controls (33).
Fibromyalgia SyndromeTwo studies in women with fibromyalgia syndrome examined whole blood methylation related to several different genes. Of these, the study investigating 112 regions within 100 genes (including NR3C1) found no group differences in methylation (46). The larger follow-up study by the same group, which included all pilot cases, investigated 12 regions within 11 genes (including NR3C1) and found one site within GCSAML, the gene encoding the germinal center–associated signaling and motility-like protein, to be hypomethylated in women with fibromyalgia compared with controls (47).
Genome-Wide StudiesNine studies used a genome-wide approach to investigate DNA methylation in FSS. Of these, five used PBMCs as a tissue, two used T cells, and two used whole blood.
Chronic Fatigue SyndromeFour studies in women with chronic fatigue syndrome provided evidence for differences in PBMC DNA methylation as compared with healthy controls. In the first of these studies, which used the HumanMethylation450 BeadChip, 1192 differentially methylated sites and 826 genes were identified (36). Although hypermethylation was the prevailing pattern in cases overall, a relatively high proportion of hypomethylation was found in genes involved in immune cell regulation as compared with genes involved in cellular components, kinase activity, and metabolic regulation. In a follow-up cohort, in which the same microarray was used, 12,608 differentially methylated loci were identified (40). Of these, 72% were hypermethylated and 5544 were annotated to genes involved in neuronal cell development and in regulating cellular and metabolic function. Another study using the Methylation EPIC BeadChip, which covers more than 850,000 sites, identified 17,296 differentially methylated sites located in 6368 genes, of which nearly all were hypomethylated in cases and linked to metabolic function and immune regulation/stress response (42). One study used a Reduced Representation Bisulfite Sequencing approach, which covered 196,172 sites, and identified 349 differentially methylated positions, of which 44% were hypermethylated in cases as contrasted with controls. These were mapped to 122 genes involved in mitochondrial and immune function. A meta-analysis of these studies was not possible because of the heterogeneity in DNA methylation analysis/data processing approaches and/or partially unavailable data. However, at the cytosine level, the three studies that used microarrays, overlapped in 83 differentially methylated probes located to 72 genes (Supplemental Digital Content 2, https://links.lww.com/PSYMED/A957). In two of the studies, a majority of these probes were hypermethylated (36,40), whereas in one of the studies, they were hypomethylated in cases as compared with controls (42). In addition, one site (denoted “cg02212836”) within the first exon region of LY86, the gene encoding lymphocyte antigen 86, was consistently hypomethylated across studies. At the gene level, the four studies overlapped in 10 genes, namely, MAD1L1, C7orf50, MEGF11, COL18A1, PLEC, TMCO3, UMODL1, RPS6KA2, NFATC1, and CD6.
Two studies investigated T cell methylation, both using the 450K HumanMethylation BeadChip. The first study found 120 differentially methylated positions in CD4+ cells, which were associated with 75 genes (35). One quarter of the identified loci were hypermethylated, whereas the rest was hypomethylated. The second study identified 15 differentially methylated sites in CD3+ cells and 31 related genes, with hypermethylation being the prevailing pattern in cases as compared with controls (41). Although a meta-analysis of these studies was not possible because of partially unavailable data, the findings overlapped regarding hypermethylated sites within HLA-DQB1, encoding the major histocompatibility complex class II antigen DQ beta 1 chain.
Fibromyalgia SyndromeTwo studies in women with fibromyalgia syndrome determined whole blood DNA methylation using the HumanMethylation450 BeadChip microarray, which covers more than 450,000 sites (34,39). The first of these studies identified 69 differentially methylated sites, of which 63 were hypermethylated in the women with fibromyalgia syndrome (34). The sites were localized to 47 different genes (including BDNF) and functional clusters involved in nervous system development and neuron differentiation. The second study identified 1610 differentially methylated sites and 960 associated genes, of which one-third was hypermethylated in the women with fibromyalgia syndrome (39). The relevant genes were linked to DNA repair, membrane transport, lipid metabolism, and the immune system. Although a meta-analysis of these studies was not possible because of partially unavailable methylation data, eight of the genes identified by the first study were replicated by the second study, namely, HDAC4, encoding histone deacetylase 4; TMEM44, encoding the transmembrane protein 44; KCNQ1, encoding the potassium voltage-gated channel subfamily Q member 1; SLC17A9, encoding the solute carrier family 17 member 9; PRKG1, encoding the CGMP-dependent protein kinase I; ALPK3, encoding alpha kinase 3; TFAP2A, encoding the transcription factor AP-2 alpha; and LY6G5C, encoding the lymphocyte antigen-6 G5C.
Irritable Bowel SyndromeOne PBMC study using the HumanMethylation450 BeadChip identified one hypermethylated site in cases as compared with controls, and this site was located to SNCAIP, the gene encoding the synuclein alpha interacting protein (38).
DISCUSSIONThe present systematic review provides tentative evidence for alterations in DNA methylation in FSS, and in relation to genes which may be relevant to symptoms such as fatigue and pain. We report two main findings: first, candidate-gene studies point toward a role of relative hypomethylation of specific sites within NR3C1 in chronic fatigue syndrome. Second, genome-wide studies in chronic fatigue syndrome, fibromyalgia syndrome, and irritable bowel syndrome identified additional sites of differential methylation within genes regulating cellular signaling and immune functioning.
The findings from candidate-gene studies fit in well with previous pathophysiological observations in FSS. More specifically, the finding of whole-blood hypomethylation related to NR3C1, encoding the glucocorticoid receptor, in chronic fatigue syndrome fits in well with hypocortisolism as a key feature of individuals with chronic fatigue syndrome and women with fibromyalgia syndrome (31), which has been demonstrated to map onto symptom exacerbations (e.g., (48,49)). Interestingly, some of the studies included in this systematic review have indeed provided direct evidence for hypomethylation of the NR3C1 promoter in chronic fatigue syndrome to be linked with increased glucocorticoid sensitivity (37) and fatigue severity (43). When taken together, this may mean that hypomethylation of the NR3C1 promoter causes increased expression of the glucocorticoid receptor, which may, in turn, fuel hypocortisolism and, ultimately perpetuate chronic fatigue syndrome (Figure 2). It is noteworthy that NR3C1 was not identified as a differentially methylated gene in genome-wide analyses in chronic fatigue syndrome. However, all existing genome-wide studies used PBMCs or T cells as tissue and are thus not directly comparable to the candidate-gene studies, which used whole blood. Moreover, despite the evidence for hypocortisolism in women with fibromyalgia syndrome (31), the studies in this population did not yield similar evidence, which could indicate that only subgroups of individuals with high levels of medically unexplained fatigue are affected by differential methylation in NR3C1. However, this is pure speculation. Further candidate-gene research replicating and extending the presented findings is urgently warranted, especially in light of the small number of existing studies and the fact that they were conducted by the same group. Furthermore, large-scale genome-wide studies in whole blood will be necessary to shed light on the relative importance of NR3C1 methylation in FSS pathophysiology. Importantly, any future studies should also include measures of gene expression, which are necessary to establish the hypothesized functional implications of the differential methylation.
 FIGURE 2:
FIGURE 2: Hypothesized associations of NR3C1 promoter hypomethylation. NR3C1 = nuclear receptor subfamily 3 group C member 1.
The findings from the genome-wide studies are also in line with prominent pathophysiological notions in FSS. The studies in chronic fatigue syndrome provided nuanced insights. In PBMCs, 83 differentially methylated sites between cases and controls were identified across studies, with both hypermethylated and hypomethylated patterns observed. One explanation for these discrepancies could be different stages of illness, such that hypermethylation at illness onset progresses into hypomethylation over time, a trajectory frequently seen regarding stress-induced alterations in hypothalamic-pituitary-adrenal axis activity (50). Given that no information on illness duration was available from the included studies, this hypothesis remains to be tested—and preferably by large-scale longitudinal studies. Interestingly, there was convergence regarding relative hypomethylation in chronic fatigue syndrome in a specific position within the first exon region of LY86, which mediates innate immune response to lipopolysaccharide and cytokine production. In T cells, there was an additional finding of relative hypermethylation in HLA-DQB1, encoding surface molecules presenting peptides from extracellular proteins. The fact that these genes were affected is concordant with the observation of low-grade inflammation in FSS (18–20,51), which may potentially develop as a result of chronic hypocortisolism (see previous section) (31). Indeed, one of the included studies in chronic fatigue syndrome found an association between hypomethylated loci governing immune function and enhanced glucocorticoid sensitivity (40). However, again, given the low number of available studies and methodological diversity, caution is advised regarding an overinterpretation of these findings, and gene expression analyses need to be included in future studies to assess the functional implications of the differential methylation. The whole-blood studies in fibromyalgia syndrome yielded eight genes that related to sites that were differentially methylated between individuals with affected individuals and controls. Among these were TMEM44, KCNQ1, SLC17A9, PRKG1, ALPK3, TFAP2A, and LY6G5C, regulating cellular signaling and immune processes. Unfortunately, methylation data were only partially available, and therefore, it remains unknown whether hypermethylation or hypomethylation was the prevailing pattern, rendering it difficult to draw any conclusions on the role of these changes in fibromyalgia syndrome pathophysiology.
The significant changes in DNA methylation in chronic fatigue syndrome and fibromyalgia syndrome are in line with the chronicity of FSS and raise the important question of whether these marks can be “edited” and whether this would be associated with symptom alleviation. One of the most promising treatments that may induce such molecular edits is psychotherapy, in particular cognitive behavioral therapy (52–54). A handful of studies in FSS have already demonstrated that cognitive behavioral therapy is associated with increases in glucocorticoid receptor-α mRNA (55) and cortisol levels (55–57). In addition, candidate-gene research in posttraumatic stress disorder, another stress-related disorder characterized by hypocortisolism, has revealed that changes in DNA methylation marks of intron 7 and exon 1 of FKBP5 were associated with positive psychotherapy outcomes (58). This gene encodes the FK506 binding protein 5, a heat shock protein 90–associated co-chaperone of glucocorticoid signaling. Finally, more recently, a genome-wide study has identified 12 methylated regions that were significantly linked with the extent that the patients responded to psychotherapy (59). These findings are promising in suggesting that DNA methylation is not only altered in response to negative environmental influences, such as stress, but may change again once the contextual conditions improve.
The present study presents with a number of strengths. It is the first systematic overview of the literature on DNA methylation in FSS, a topic of particular importance given that affected individuals often face stigmatization due to the medically unexplained nature of their complaints and given the need for a more in-depth pathophysiological understanding of these illnesses. The study also followed well-established guidelines (e.g., MOOSE) and included a quality assessment/risk of bias rating. Although the score of 5 of 10 quality points may seem modest at first, this was largely driven by missing information on the blinding of laboratory staff regarding participant group status. Although we still feel it is important for the authors of future studies to explicitly mention this, we also acknowledge that, in reality, DNA methylation analyses are mostly implemented on a commercial basis, with independent laboratories operating under blinded conditions. In combination with the fact that the included studies provided excellent descriptions of DNA methylation analyses and considered important confounders, such as age, sex, and body mass index, the risk of bias was considered low to moderate overall. Finally, another strength of the candidate-gene studies, which was not formally assessed in this systematic review, was that all of them provided clear rationales for their choice of candidate genes. Nevertheless, a number of limitations are also worth mentioning. First, the sample sizes of the included studies were relatively small, and the lack of available data mostly prevented us from being able to undertake meta-analysis. Future, large-scale research providing access to summary statistics of all sites investigated is thus warranted. Second, several of the studies did not exclude comorbid conditions, the intake of medication, and smoking, which could have impacted on DNA methylation, and future research would certainly benefit from at least a more thorough documentation of these variables. Third, the included studies used whole blood, PBMCs, or T cells as tissue to determine DNA methylation. This not only means that the herein obtained findings do not necessarily translate to other cell types, but also meant that findings from candidate-gene studies (which were mostly conducted in whole blood) and the findings from genome-wide studies (which were mostly conducted in PBMCs) were difficult to compare. Finally, all but one of the included genome-wide studies relied on gene ontology and network analyses to interpret their findings, without any direct measure of gene expression. Therefore, the functional implications (and hence the clinical relevance) of the majority of the identified differentially methylated positions in FSS have yet to be proven.
To conclude, there is tentative evidence in favor of altered DNA methylation in chronic fatigue syndrome and in fibromyalgia syndrome. In chronic fatigue syndrome in particular, there is evidence that these alterations are linked to pathways that may bear relevance to key pathophysiological alterations (i.e., hypocortisolism and low-g
留言 (0)