4.1 Plasma Pharmacokinetics of GalNAc-Conjugated siRNAs
We collected and analyzed literature pharmacokinetic data for nine GalNAc-conjugated siRNAs. Based on this analysis, we showed that the clinical plasma AUC and Cmax of GalNAc-conjugated siRNAs are approximately dose proportional and similar between the chemical stabilizing methods used. The reported linear regression equations for AUC and Cmax, in combination with the approximately twofold uncertainty, can be useful to predict and reason about plasma pharmacokinetics for other GalNAc-conjugated siRNAs.
To support the design and interpretation of animal studies and to reason about translation, we collected corresponding plasma pharmacokinetics and toxicokinetic data from rodents and monkeys. Like human data, animal data indicate dose-proportional Cmax and similar kinetics between compounds with different chemical stabilizing methods. However, in contrast to human data, animal data show supra-dose-proportional AUCs. Our observation that plasma AUC is approximately dose proportional in humans at therapeutic dose levels, and greater than dose proportional in monkeys for a wider dose range up to 300 mg/kg, agrees with data reported by McDougall et al. [9] for seven GalNAc-siRNAs with unknown targets and sequences. The supra-proportionality observed when including higher dose levels is likely due to transient saturation of the ASGPR, which takes up the GalNAc-siRNA to the hepatocytes [9]. The dose ranges differ between animals and humans because the animal studies contain data from toxicity studies at dose levels up to 300 mg/kg. It is likely that human data would also show supra-proportionality if studied at greater than therapeutic dose levels. We note that awareness of potential non-linearities in pharmacokinetics is critical to correctly interpret how high-dose levels impact pharmacodynamics.
Translation between species follows allometric principles with a clearance over bioavailability exponent of 0.75. The estimate is based on an analysis of all data simultaneously, but a similar result is obtained when each compound is analyzed individually. The value aligns well to the often-assumed slope of 0.75, sometimes referred to as Kleiber’s law [33], which also has empirical support for various drug classes, including oligonucleotides, e.g., Jansen et al., Oitate et al., and Geary et al. [34,35,36].
Although plasma pharmacokinetics are similar for various GalNAc-siRNAs, the target tissue half-life may differ substantially (1.5–14 weeks in humans) [6]. Therefore, the dose level and dosing frequency of in vivo efficacy studies are mainly determined by biomarker response data over weeks, and not by plasma pharmacokinetic data sampled over only 24–48 h. Nevertheless, plasma pharmacokinetics are critical for reasoning on safety windows and on how non-linearities in pharmacokinetics may affect pharmacodynamics. Moreover, plasma pharmacokinetics observed over weeks, requiring high-sensitivity bioanalysis methods, would likely be useful for both efficacy and safety reasoning.
4.2 Pharmacokinetic Models for GalNAc-Conjugated siRNAs
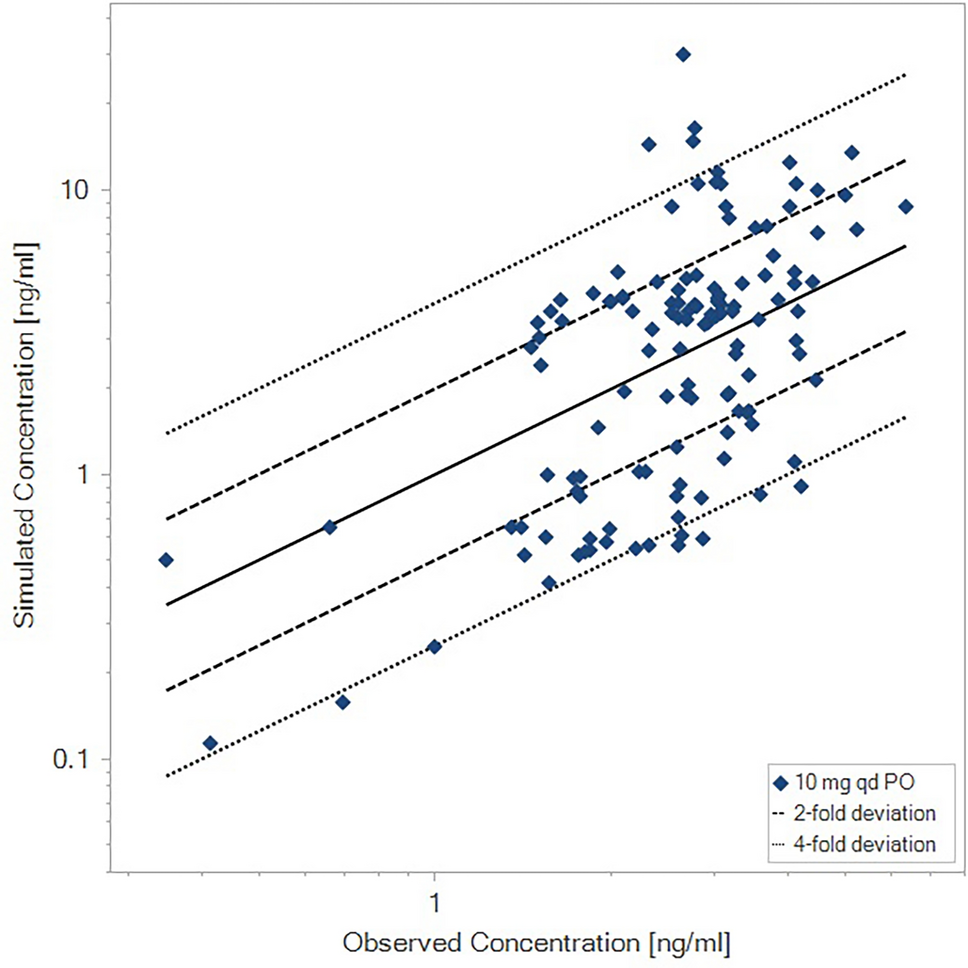
Clinical plasma concentration-time profiles of GalNAc-siRNAs can be described by one-compartment kinetics with first-order absorption over the first 24 h, and by three-compartment kinetics for the rare case when additional later time-points up to several weeks after dosing were sampled and exposure detected. The reported models can be useful in the preclinical phase of new GalNAc-siRNAs, e.g., to reason about safety windows. When the first pharmacokinetic data have been generated in phase I, it is natural to develop a clinical pharmacokinetic model specifically to the GalNAc-siRNAs under consideration. The three-compartment models, based on data from either olpasiran alone or five GalNAc siRNAs, differ noticeably in several model parameters, including absorption rate (Ka). This is somewhat in agreement with the estimated range of Ka values for the one-compartment models (ESM Table S4) and should also be viewed in relation to the moderately large standard errors for the parameters in the olpasiran model. In contrast, the model based on all data features much smaller standard errors for all parameters and appears more robust. However, some caution is warranted to avoid overinterpreting, specifically those parameters in which the two models differ.
An alternative to compartmental models is to develop a mechanistic model allowing a broader range of scenarios to be tested, e.g., simulations of tissue pharmacokinetics and GalNAc uptake. We exemplify this type of model using the reported PBPK model by Ayyar et al. [30]. The usefulness of this model is limited by two factors. First, the model code and raw data are not included in the publication, and the reported equations and parameters fail to reproduce the simulations in the published paper. Second, the model is not trained on plasma pharmacokinetic data beyond 48 h and fails to describe such data for olpasiran. By identifying and correcting several errors in the published model equations, we managed to recreate, as faithfully as possible, the model simulations reported by Ayyar et al. [30]. Despite its limitations, we believe the PBPK model can be a good starting point for improved mechanistic models of the pharmacokinetics of GalNAc-siRNAs, and we report our corrected model to facilitate such extensions (see the ESM).
Today, when reasoning about the clinical plasma pharmacokinetics in the preclinical phase of a new GalNAc-siRNA, we argue that our proposed three-compartment models (Table 3) constitute good model choices when simulations over weeks are desired. In comparison with the reported PBPK model, they more accurately describe the terminal plasma half-life. For simulations over 24–48 h, any of the reported simpler one-compartment models are adequate.
In general, both simple (e.g., allometric and compartmental) and complex (e.g., PBPK) models have their advantages and disadvantages. Briefly, the relevant model complexity depends on (1) data quality and quantity; (2) prior information of the system; and (3) the research question to be addressed. Because of this, it is common and natural that several models, defined at different levels of complexity, exist in parallel. Models of the type presented here can help to address several research questions in the discovery phase. Overall, they can support decision making all the way from target selection to candidate selection. Specifically, they can inform the choice of type of drug class (e.g., small molecule, peptide, or siRNA) that is adequate from an efficacy and safety perspective to hit a certain target, they can help to guide the design of in vivo experiments, and anchoring reasoning about the translational aspects of a new candidate compound, from both a pharmacokinetic/pharmacodynamic perspective and a toxicokinetic-to-safety perspective. In this context, the benefits of a simple model, compared with a complex PBPK model, include simple to implement, simple to use, and simple to unambiguously define. The benefits of a PBPK model include mechanistic insight, e.g., considering species differences of both receptor density and the receptor turnover rate of ASGPR, and the much wider range of possible simulations. Naturally, the value of a certain model, independently of model complexity, increases if it has been shown that the pharmacokinetics of the drug class are similar between different chemical modifications, something that we have shown here for GalNAc-siRNAs.
4.3 Limitations of the Analysis
We have identified the following main limitations of our analysis. First, the number of investigated GalNAc-siRNAs is limited; nine for humans, seven for monkeys, and five for rats. This reflects the relatively low number of GalNAc-siRNAs with clinical pharmacokinetic data in the public domain, although the numbers are expected to gradually increase in the coming years. On the other hand, there is relatively low spread in our AUC and Cmax regressions, with approximately twofold uncertainty around the point predictions. Second, our analysis uses mean or median data and not individual data; therefore, any prediction using our models should be interpreted on the group level and not on the individual level. Third, the group size for the animal studies was not always reported and an assumption of three subjects per group was made when the information was missing. However, similar parameters were obtained when repeating the regression without weighting (see the ESM), indicating that the sensitivity to this assumption is at least not severe. Fourth, the rat analysis was performed on compounds only representing Alnylam’s chemical stabilizing method. However, our data indicate no dependency between plasma pharmacokinetics and the chemical stabilizing method in monkeys and humans, and it is therefore likely that the same holds in rats. Fifth, one cannot exclude some variability in accuracy and precision between the various bioanalytical methods used for the different compounds (ESM Table S1).


留言 (0)