Animals
We obtained adult male Sprague–Dawley (SD) rats weighing 260–280 g from Shanghai Laboratory Animal Research Center (Shanghai, China). Only male rats were used in this study because a previous study documented the neuroprotective effects of oestrogens on animal models of cerebral ischemia [53]. We housed all rats under standard laboratory conditions of of 23 ± 2 °C, 40–50% humidity, and 12 h–12 h light-dark cycle. Food and water were freely available to the animals. The Animal Ethics Committee of Shanghai University of Traditional Chinese Medicine reviewed and approved all experimental protocols, and all animal procedures followed the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Animal model of ischemic stroke and intracerebroventricular injection
Pentobarbital sodium (30 mg/kg, intraperitoneal) was used to anesthetize rats. Then, rat was placed in a supine position, and a midline incision was made on the neck to expose the left external carotid artery (ECA), internal carotid artery (ICA) and common carotid artery (CCA). Surgical silk was used for ligation of the ECA to block blood flow, and the left ICA and CCA were temporarily clamped using microvascular clips. In the ECA, a small incision was made. Then, a silicon-coated suture (L3600, Jia Ling Biotechnology Co. Ltd., Guangzhou, China) was gently advanced from the ECA stump into the ICA to occlude blood flow in the MCA. The silicon-coated monofilament nylon suture was fixed for 2 h, and ICA perfusion was restored. Rats in the sham group underwent the same operation with the exception of the monofilament nylon suture being inserted.
The animals were randomly assigned to 4 groups: the sham group, MCAO/R group, MCAO/R + agomir-NC group and MCAO/R + agomir-212-5p group. According to previous reports, agomir-212-5p and agomir-NC were injected intracerebroventricular (ICV) injection 30 min before MCAO/R [54]. The skull was exposed by making an incision along the midline of the scalp. Injection coordinates were 1.0 mm posterior and 1.5 mm lateral to the bregma at a depth of 3.5 mm. Agomir-212-5p or agomir-NC (10 µM in 7 µl) was slowly injected. Simultaneously, 7 µl of 0.9% NaCl was injected into lateral ventricle both in the sham and MCAO/R groups. The needle was retained in situ for 5 min following injection.
Isolation of microglial exosomes from the injured brain
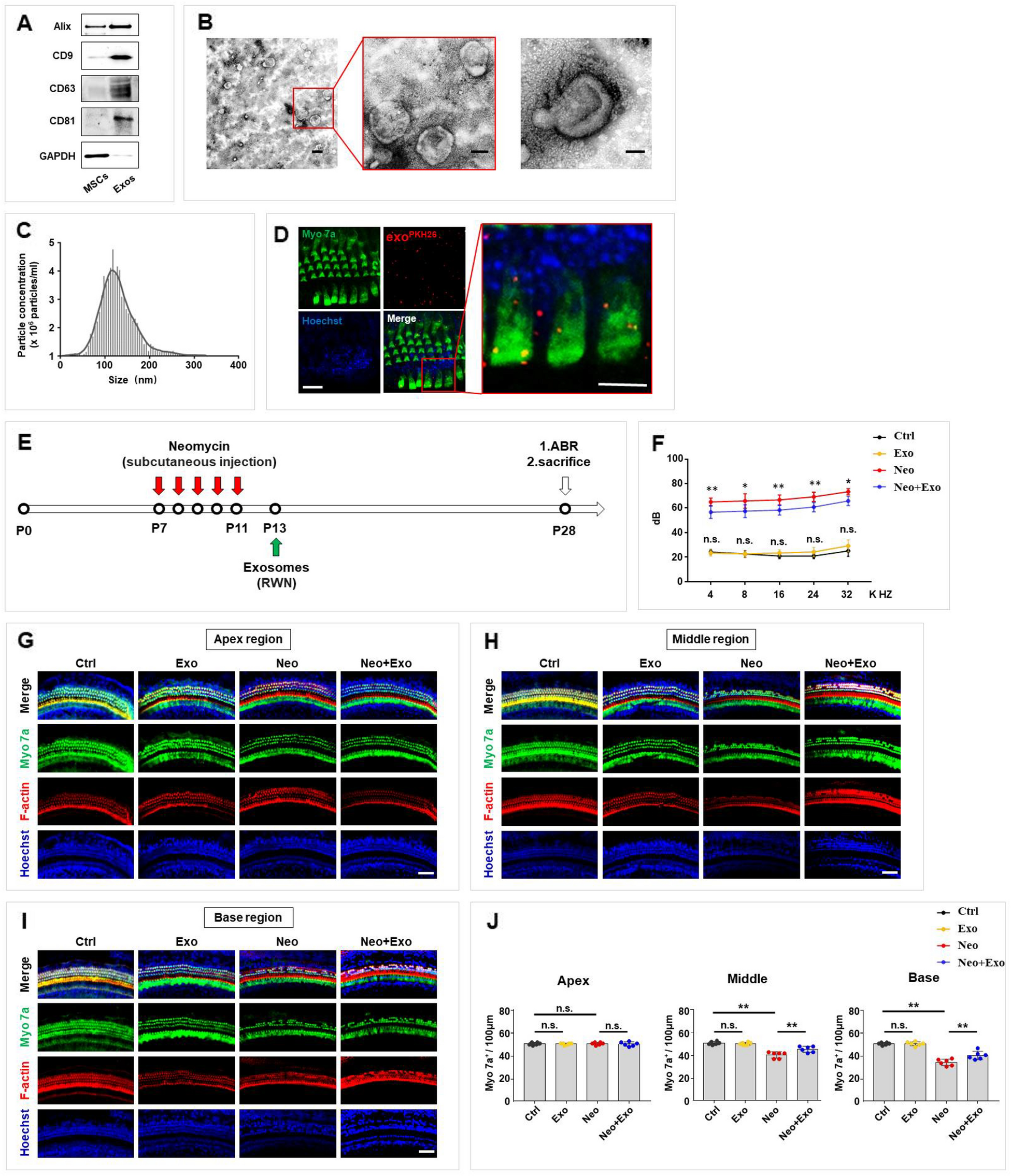
Microglial exosomes from the ischemic penumbra portion of the cortex were isolated as previously described [36]. In each group, the ischemic penumbra portion of the cortex was rapidly isolated from the left hemisphere. Samples were digested with collagenase type 3 (75 U/ml, Worthington Biochemical Corporation, Lakewood, NJ, USA) at 37 °C for 15 min. Then, the cells were centrifuged at 2000 × g at 4 °C for 10 min to remove dead cells and subsequently centrifuged again for 30 min at 10,000 × g at 4 °C to remove cellular debris. After collecting the supernatant, it was filtered through a 0.22 μm filter. Samples were centrifuged at 100,000 × g at 4 °C for 70 min, and the supernatant was removed. Calcium- and magnesium-free Dulbecco’s PBS (Gibco, USA) was applied to resuspend the exosome pellet. Samples were incubated for 1 h with 50 mL of 3% BSA containing 1.5 µg of anti-CD11b biotinylated antibody (NB110-89474B, Novus, CO, USA) at room temperature to isolate microglial exosomes. Subsequently, the mixture was incubated with Pierce Streptavidin Plus UltraLink Resin (Thermo Fisher Scientific, Waltham, MA, USA) at room temperature for 30 min. Thereafter, centrifuged at 800 × g for 10 min at 4 °C of the samples was performed. The supernatant was discarded after centrifugation, and microglial exosomes were recovered and stored at 4 °C until use in subsequent analysis.
Exosome identification
Nanoparticle tracking analysis (NTA), transmission electron microscopy (TEM) and western blot analysis were applied to characterize exosomes. First, exosomes were analysed by performing NTA. In order to measure the size and concentration of exosome particles, we used NTA ZetaView PMX 110 (Particle Metrix, Meerbusch, Germany) and ZetaView 8.04.02. Isolated exosome samples were appropriately diluted in 1 × PBS (Biological Industries, Israel). NTA was performed at 11 positions, and the measurement results were recorded. The ZetaView system was calibrated using 110 nm polystyrene particles. Temperature was maintained at approximately 26.26 and 27.21 °C.
Exosome morphology was analysed using negative-staining TEM. In 50 µl of 2% PFA, exosomes were resuspended, and the exosomal suspension (5 µl) was placed on Formvar/carbon-loaded copper mesh and washed 3 times in PBS. The copper mesh was floated onto a drop of 1% glutaraldehyde for 5 min and then washed. After washes with distilled water, copper meshes were placed on droplets of a uranyl-oxalate solution for 5 min and then transferred to methylcellulose for 10 min. Excess liquid was removed with filter paper and samples were allowed to air dry for 10 min. Electron micrographs were obtained with a JEOL 1230 transmission electron microscope (JEOL, Tokyo, Japan).
The expression of the exosome marker proteins CD9, CD63 and CD81 was evaluated using western blot analysis. To identify microglia-derived exosomes, CD11b was used. Exosome samples were first lysed using RIPA buffer, and total protein was separated on 12% SDS–PAGE gels and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA). After blocking the membranes for 1 h with 10% skim milk formulated in Tris-HCI buffered saline solution (TBST), they were overnight incubated with primary antibodies. Antibodies included CD9 (1:1000, ab92726, Abcam, Cambridge, UK), CD63 (1:1000, AF5117, Affinity Biosciences, Cincinnati, OH, USA), CD81 (1:500, DF2306, Affinity Biosciences, Cincinnati, OH, USA) and ITGAM/CD11b (1:500, DF2911, Affinity Biosciences, Cincinnati, OH, USA). The PVDF membranes were then incubated with HRP-conjugated secondary antibodies (1:5000) at room temperature for 1 h. UVP BioSpectrum imaging system (BioSpectrum 410, USA) was used to detect protein signals using WesternBright ECL reagent (Advansta, USA).
MiRNA sequencing analysis and target gene prediction
MiRNA sequencing was performed by KangCheng Biotech, Shanghai, China. After the total RNA samples were extracted with TRIzol reagent and tested and quantified using agarose gel electrophoresis and a NanoDrop spectrophotometer, the library was constructed, and the quality of the library was tested using an Agilent 2100 Bioanalyzer. In order to generate single-stranded DNA, mixed sequencing libraries of different samples were denatured with 0.1 M NaOH, and 51 cycles of sequencing were performed using an Illumina NextSeq 500 sequencer according to the supplier’s instructions. TargetScan (http://targetscan.org/) and miRDB (http://mirdb.org/miRDB/) were used to predict miR-212-5p downstream target genes, and genes related to neurodegeneration were selected for further analysis.
Dual-luciferase assay
The wild-type PLXNA2 3′UTR (WT) or PLXNA2 3′UTR mutant (MUT) of the miR-212-5p binding site, was inserted into the dual-luciferase pmirGLO vector (GenePharma, Shanghai, China). The WT and MUT luciferase reporter plasmids were cotransfected into PC12 cells along with miR-212-5p or negative control (NC) using Lipofectamine 2000 (GenePharma Co., Ltd., Shanghai, China) according to the manufacturer’s protocol. Luminescence was detected with a dual-luciferase reporter assay (Promega, Madison, WI, USA) using a GloMax instrument (Promega).
Cell culture, treatments, and oxygen–glucose deprivation/reperfusion (OGD/R)
We obtained PC12 cells from the Shanghai Institute of Cell Biology and cultured them in DMEM containing 10% FBS and 1% penicillin/streptomycin. The cells were cultured at 37 °C in a humidified 5% CO2 environment. For experiments with transfected cells, cells were transfected with 100 nM agomir-212-5p or agomir-NC. Transfected cells were used for subsequent experiments 24 h after transfection. We then changed the culture medium to glucose-free culture medium. The cells were incubated at 37 °C in a low-oxygen incubator for 4 h (1% O2, 94% N2, and 5% CO2). Subsequently, the cells were switched to regular medium and maintained under standard (5% CO2 and 37 °C) conditions for reperfusion.
Behavioral tests
Neurological deficits were scored by an observer blinded to the groups using Zea Longa scores [55]: 0 = no symptoms of neurological deficits; 1 = cannot extend the right forepaw completely; 2 = circling to the right; 3 = falling to the right; 4 = unable to walk spontaneously.
The foot fault test was performed in accordance with a previously described method [56]. Eight rats were included in each group. We tested rats’ ability to walk over a ladder with irregular spacing (1–3 cm) on 3 occasions and video recorded to assess the impairment in right forelimb function after stroke. Scoring was based on the following criteria: 0 = total miss; 1 = deep slip; 2 = slight slip; 3 = replacement; 4 = correction; 5 = partial placement; 6 = correct placement. The misstep rate refers to the number of steps with a walking score of 0, 1 or 2. To calculate the misstep rate, we used the following formula: (the number of wrong steps of the right forelimb/total steps) × 100%.
The motor performance and coordination of animals were evaluated using an automated quantitative gait analysis system (CatWalk™, Wageningen, The Netherlands). Each group included eight rats. During the experiment, the room was dark and quiet. Prior to experimentation, each rat performed 3 trials without any interruption to cross the pressure-sensitive plate of the Catwalk system. The rat was placed at one end of a 150-cm-long runway consisting of a glass platform covered by a black tunnel with a food reward at the opposite end of the runway. This test was performed before and on days 1, 3, 5, and 7 following the surgery, respectively. Data were acquired and analysed using CatWalk version 10.6 software.
Motor evoked potential (MEP)
Electrophysiological tests were conducted on the 7th day following modelling with an electromyography evoked potentiometer (9033A07, Keypoint; Medronic, Skovlunde, Denmark). After the rat was anesthetized, the recording electrode was placed on the right biceps brachii, and the stimulating electrode was inserted into the rat’s upper jaw which was near the left motor cortex. Single square-wave electrical pulse (100 µs) was applied, and the stimulation intensity increased gradually until latency and amplitude were no longer changed. The latency and amplitude of the MEP were obtained.
Resting-state functional magnetic resonance imaging (fMRI)
We used a 11.7 T scanner (Bruker, Ettlingen, Germany) for the fMRI scans. 5% isoflurane was used to induce anesthesia, and anesthesia was maintained with isoflurane (1.5%) and dexmedetomidine (0.05 mg/kg). Resting-state fMRI data were collected using single echo planar imaging (EPI) with the following parameters: flip angle = 90°, slice thickness = 0.3 mm, number of average = 1, repetition time (TR) = 3000ms, Echo time (TE) = 8.142 ms, field of vision (FOV) = 27 × 27 mm2. The fMRI data were preprocessed using Statistical Parametric Mapping 8 (SPM 8) toolbox (http://www.fil.ion.ucl.ac.uk/spm/) based on the MATLAB (R2014b; Mathworks, Natick, MA, USA). All the images were transformed to Nifti format, followed by slice timing and realign. Non-brain tissues were removed manually and each image was manually reoriented by setting the origin to the anterior commissure. Finally, images processing with normalize and smooth. In the following FC analysis, the ipsilateral (left) motor cortex was selected as the region of interest (ROI) to examine whole-brain functional connectivity, and then performing Fisher’s Z transformation. The comparison between groups was carried out using two-sample t-test in SPM8. P < 0.001 was considered to be statistically significant.
Evaluation of the infarct volume
Rat brains were quickly removed and frozen at -20 °C for 20 min; subsequently, the frozen brains were coronally sliced into five 2-mm-thick coronal slices. Brain tissue slices were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma-Aldrich, St. Louis, MO, USA) and incubated at 37 °C for 20 min in the dark, followed by fixation with 4% paraformaldehyde (PFA). The infarct area was calculated using Image J software.
Western blot analysis
Brain tissue corresponding to the ischemic penumbra and cells were collected and homogenised with RIPA lysis buffer (Beyotime, Shanghai, China). By using BCA protein assay reagents (Beyotime, Shanghai, China), protein concentrations were determined. A tissue sample (80 µg) and cellular sample (30 µg) were loaded and separated on SDS–PAGE gels prior to being transferred to a PVDF membrane. The PVDF membranes were blocked with protein-free rapid blocking buffer for 20 min and then incubated with primary antibodies against PLXNA2 (1:1000, #5658, Cell Signaling Technology, Beverly, MA, USA), Rho protein A (RhoA) (1:5000, ab187027, Abcam, Cambridge, UK), Rho-associated kinases 2 (ROCK2) (1:10000, ab125025, Abcam, Cambridge, UK), GAP43 (1:100000, ab75810, Abcam, Cambridge, UK), neurite outgrowth inhibitor-A (Nogo-A, 1:1000, DF8581, Affinity Biosciences, Cincinnati, OH, USA), nogo receptor (NgR, 1:500 DF13593, Affinity Biosciences, Cincinnati, OH, USA), and β-actin (1:1000, #4970, Cell Signaling Technology, Beverly, MA, USA) at 4 °C overnight. On the following day, the membranes were incubated with HRP-conjugated secondary antibodies (1:5000) for 1 h at room temperature. The UVP BioSpectrum imaging system (BioSpectrum 410, USA) was used in conjunction with WesternBright ECL reagent (Advansta, USA) in order to detect protein signals. Finally, all bands were subjected to densitometry analysis with ImageJ.
H&E and nissl staining
For Nissl staining, haematoxylin–eosin (H&E) staining and immunofluorescence staining, samples of brain were fixated in 4% paraformaldehyde, then dehydrated and embedded in paraffin. Subsequently, brain Sect. (5 μm thick) were fixed on poly-L-lysine-coated slides for H&E staining and Nissl staining. For HE staining, the slices were stained with haematoxylin and eosin with an H&E assay kit (G1003, Servicebio, Wuhan, China). Slices were stained with 0.1% cresyl violet for Nissl staining (G1036, Servicebio).
Immunofluorescence staining
For paraffin tissue sections, sections were dewaxed with xylene and rehydrated with alcohol. The sections were subjected to heat-induced antigen retrieval, blocked with 10% goat serum albumin containing 0.3% Triton X-100, and incubated with the indicated primary antibodies overnight at 4°C. PC12 cells were washed with PBS, fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton X-100 in PBS for 15 min, and incubated with 10% goat serum albumin at room temperature for 1 h. Next, slides were incubated at 4°C overnight with primary antibodies against Iba1 (1:100, ab15690, Abcam, Cambridge, UK), CD206 (1:200, ab64693,, Abcam, Cambridge, UK), CD86 (1:200, PA5-88284, ThermoFisher, Waltham, MA, USA), PLXNA2 (1:200), RhoA (1:150), ROCK2 (1:200), NeuN (1:200, ab104224, Abcam, Cambridge, UK), Cleaved-Caspase 3 (1:200, BF0711, Affinity Biosciences, Cincinnati, OH, USA), Nogo-A (1:200), NgR (1:200), microtubule-associated protein 2 (MAP-2, 1:200, 17490-1-AP, Proteintech, Wuhan, China) and β III tubulin (1:500, ab52623, Abcam, Cambridge, UK). The following day, the samples were incubated with Alexa Fluor® 488-conjugated AffiniPure goat anti-rabbit (1:200, 33106ES60, YEASEN, Shanghai, China) or Alexa Fluor® 594-conjugated AffiniPure goat anti-mouse (1:200, 33212ES60, YEASEN, Shanghai, China) secondary antibodies at 37°C for 30 min. Nuclear staining was performed using 4’, 6-Diamidino-2-phenylindole (DAPI, Beyotime, Shanghai, China). All images of immunofluorescence staining were captured using a fluorescence microscope (DM6000B, Leica, Germany). Images were analysed using ImageJ software.
Quantitative RT–PCR (qRT–PCR)
Total RNA samples from cells and brain tissues using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and were reversely transcribed to cDNA using reverse transcription kit (A3500, Promega, Madison, WI, USA). Target genes were examined using a SYBR-Green RT–PCR kit (QPK-212, TOYOBO, Osaka, Japan) and a LightCycler 480 system (Roche, San Francisco, CA, USA). PCR conditions were as follows: predenaturation for 5 min at 95 °C, followed by 40 cycles of 95 °C for 10 s, 60 °C for 10 s, and 72 °C for 10 s. U6 (for miRNA) and β-actin (for mRNA) were used as an internal inference. Reactions were performed in triplicate, and the 2–ΔΔCt method was applied to estimate the results [57]. Tables 2 and 3 show the primer sequences designed for this study.
Table 2 Primers sequence for quantitative real-time Polymerase Chain Reaction
Table 3 Results of functional connectivity analysis among four groups
Transmission electron microscopy
After cutting brain tissues into 1mm3 pieces, we fixed them with 2.5% glutaraldehyde for 2 h. The tissues were then washed and fixed in osmic acid (1%) for 1 h, dehydrated in ethanol, and embedded. Then, 50-nm ultrathin sections were prepared and stained with uranyl acetate and lead citrate. The images were captured using a transmission electron microscope (Tecnai G2 Spirit Bio TWIN, FEI Company, USA).
Golgi-Cox staining, sholl analysis and measurement of the spine density
The rat brain was dissected and subjected to Golgi-Cox staining with an FD Rapid GolgiStain Kit (FD Neurotechnologies, Columbia, MD). Rats were deeply anaesthetized with sodium pentobarbital, and brains were removed as quickly as possible while handling carefully avoiding damage to the brain tissue. Briefly, the extracted brains were soaked in a mixture of a 1:1 volumetric ratio of solutions A:B for 2 weeks in the dark at room temperature. Then, the brain tissue was transferred into another solution (C) and stored in the dark for 3 days. Coronal slices (100 μm thickness) were prepared using a vibrating slicer (Leica, VT1200 S) and stained using standard staining procedures. For the Sholl analysis, NeuronJ plugin was used for neuronal tracing and Sholl Analysis plugin (http://fiji.sc/Sholl_Analysis) was used in ImageJ. The cell body was selected, and a count of dendrite intersections around the center of the cell body was performed at 20-µm intervals. The number of spines on segments of 10 μm dendrites was counted to evaluate the dendritic spine density.
Statistical analysis
An analysis of the data was performed with SPSS Statistics software (version 22; SPSS, Chicago, IL, USA). We presented our data as mean ± standard errors of the means (SEM). The independent sample t test was used to detect significant differences between two groups. For comparisons between multiple groups, one-way analysis of variance (ANOVA) was performed. An additional post-hoc comparison was made using the least significant difference (LSD) test in the case of equal variances and Dunnett’s T3 in the case of unequal variances. It was considered statistically significant when the P was less than 0.05.








留言 (0)