Study design and patient selection
This was an open-label, phase 1/1b study of fruquintinib that enrolled U.S. patients with advanced solid tumors of any type (except squamous cell non-small cell lung cancer) conducted at 9 study centers in the U.S. In the dose escalation phase, eligible patients were adults ≥ 18 years old with histologically or cytologically confirmed, locally advanced or metastatic solid tumors who had progressed on approved systemic therapy and for which no effective treatment or standard of care was available. Additional key inclusion criteria included body weight ≥ 40 kg; at least 1 measurable target lesion according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 per investigator assessment; Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1; and adequate hematologic, hepatic, and renal function. Key exclusion criteria included prior use of a VEGF inhibitor; systemic antineoplastic therapy or any investigational therapy within 4 weeks or 5 half-lives (whichever was shorter) of the first dose of study drug; uncontrolled hypertension; history of recent bleeding; history of gastrointestinal perforation or fistula; active cardiovascular disease; or recent thromboembolic events.
This study was conducted in accordance with the protocol, the ethical principles derived from international guidelines including the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical Guidelines, applicable International Council for Harmonisation of Technical Requirements for Pharmaceuticals of Human Use (ICH) Good Clinical Practice (GCP) and other guidelines, and applicable laws and regulations. Informed consent was obtained before the patient was entered into the study.
Dose escalation and dose expansion
In the dose escalation phase, fruquintinib was administered sequentially in 2 successive cohorts, 3 and 5 mg PO QD on a 3/1 schedule on a 28-day cycle in a standard 3 + 3 design until the criteria for the MTD/RP2D were met. Dose-limiting toxicity (DLT)-evaluable patients must have met the following criteria: no previous anticancer therapy prior to DLT; completion of the first 28-day cycle with complete safety evaluation and reception of at least 85% of the assigned fruquintinib dose; or had a confirmed DLT during the first 28-day treatment cycle. If no more than 1 DLT occurred in the DLT observation window (Days 1–28 in Cycle 1 [C1]) among the 6 patients who received the 3 mg dose, DLT-evaluable patients were to be enrolled at the 5 mg dose level. If ≤ 1 patient experienced a DLT at the 5 mg dose level, then that dose level was declared the MTD/RP2D, and an additional 6 patients with solid tumors of any type were enrolled at the RP2D in the dose expansion phase (Cohort A). DLT assessment was only conducted for patients in the dose escalation phase.
A DLT was defined as any grade 4 non-hematologic adverse event (AE); any grade 3 non-hematologic AE related to study drug with the exception of nausea/vomiting, diarrhea, constipation, hypertension, and electrolyte imbalances downgraded within 3 days with appropriate supportive treatment; grade 4 neutropenia lasting > 3 days; grade 3 febrile neutropenia; grade 4 thrombocytopenia or grade 3 thrombocytopenia associated with bleeding; or dose interruption for > 14 days due to toxicity. The MTD was defined as the highest dose at which no more than 1 of 6 patients experienced a DLT.
Upon completion of dose escalation at the 3 and 5 mg dose levels and confirmation of the RP2D based on aggregated safety and PK data, additional patients were recruited in the expansion phase (Cohort A) at the RP2D to further characterize safety, tolerability, PK, and signals of clinical activity in patients with refractory solid tumors. Patients continued treatment until disease progression or any unacceptable toxicity, investigator decision to terminate therapy, or consent withdrawal.
Safety assessments
Safety was assessed by evaluation of AEs, serious AEs (SAEs), AEs of special interest (AESIs), physical examinations, vital signs, single 12-lead electrocardiograms (ECGs) and cardiac monitoring, clinical laboratory data, and ECOG performance. AEs were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) v4.03. Safety was assessed from the date of first study drug administration to 37 days after the date of last study drug administration.
Pharmacokinetic evaluations
Blood samples for determination of fruquintinib plasma concentrations were collected at pre-dose and 1, 2, 4, 8, and 24 h after dosing on Days 1, 14, and 21 of C1. The 24-h samples were collected prior to the next dose. PK samples were analyzed by LabCorp Bioanalytical (Shanghai, China) using a validated, specific, and sensitive liquid chromatography with tandem mass spectrometry assay method with an analytical range of 1.00 ng/mL (lower limit of quantitation) to 750 ng/mL (upper limit of quantitation). Inter-run variability was ≤ 5.2%. Quality control (QC) samples at 5 concentrations (3, 30, 45, 300, and 600 ng/mL) were assayed along with study samples, and the calculated QC sample concentrations deviated by ± 3.3% from the nominal concentrations.
PK parameters were determined by noncompartmental analysis using Phoenix® WinNonlin® v8.2. Systemic exposure to fruquintinib was evaluated based on PK parameters including maximum observed concentration (Cmax), time to Cmax (Tmax), and area under the concentration curve from time 0 to 24 h post-dose (AUC0-24). Accumulation ratios (AR) for Cmax and AUC were estimated from individual data and calculated as Day 14/Day 1 or Day 21/Day 1. The 3 mg dose cohort Cmax and AUC0-24 values were dose-normalized to 5 mg for inclusion in box plots. Weight-normalized apparent clearance after oral administration (CL/F) was calculated as dose/AUC0-24 divided by body weight.
Efficacy assessments
Tumor response was assessed according to RECIST v1.1 after every cycle (approximately 4 weeks) for the first 3 cycles and then every 8 weeks (± 7 days) thereafter. Assessments included computed tomography (CT) scans with oral or intravenous contrast (unless contraindicated) of the chest, abdomen, and pelvis or magnetic resonance imaging scans if CT contrast was contraindicated. Other assessment methods were used if clinically indicated. The assessment method used for each patient at baseline was the same throughout the study.
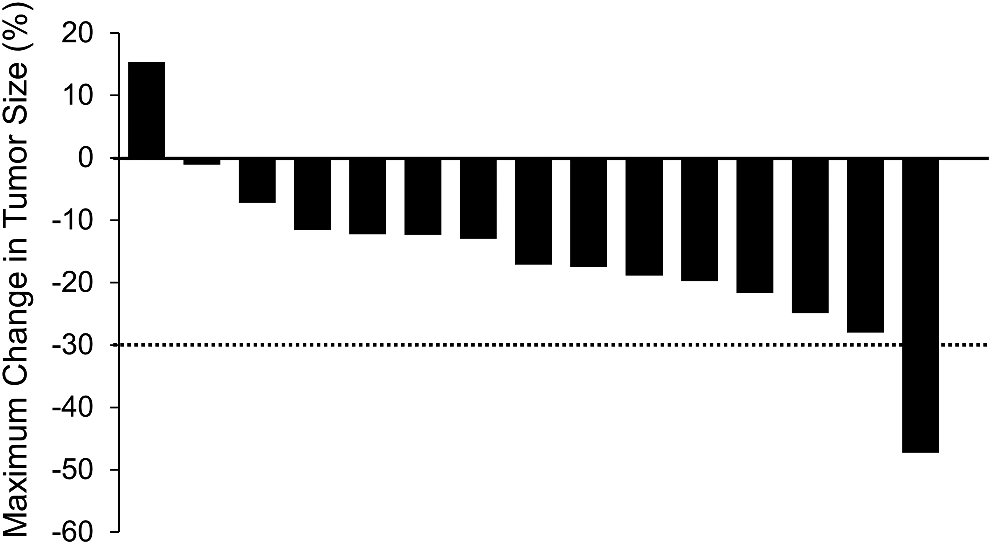
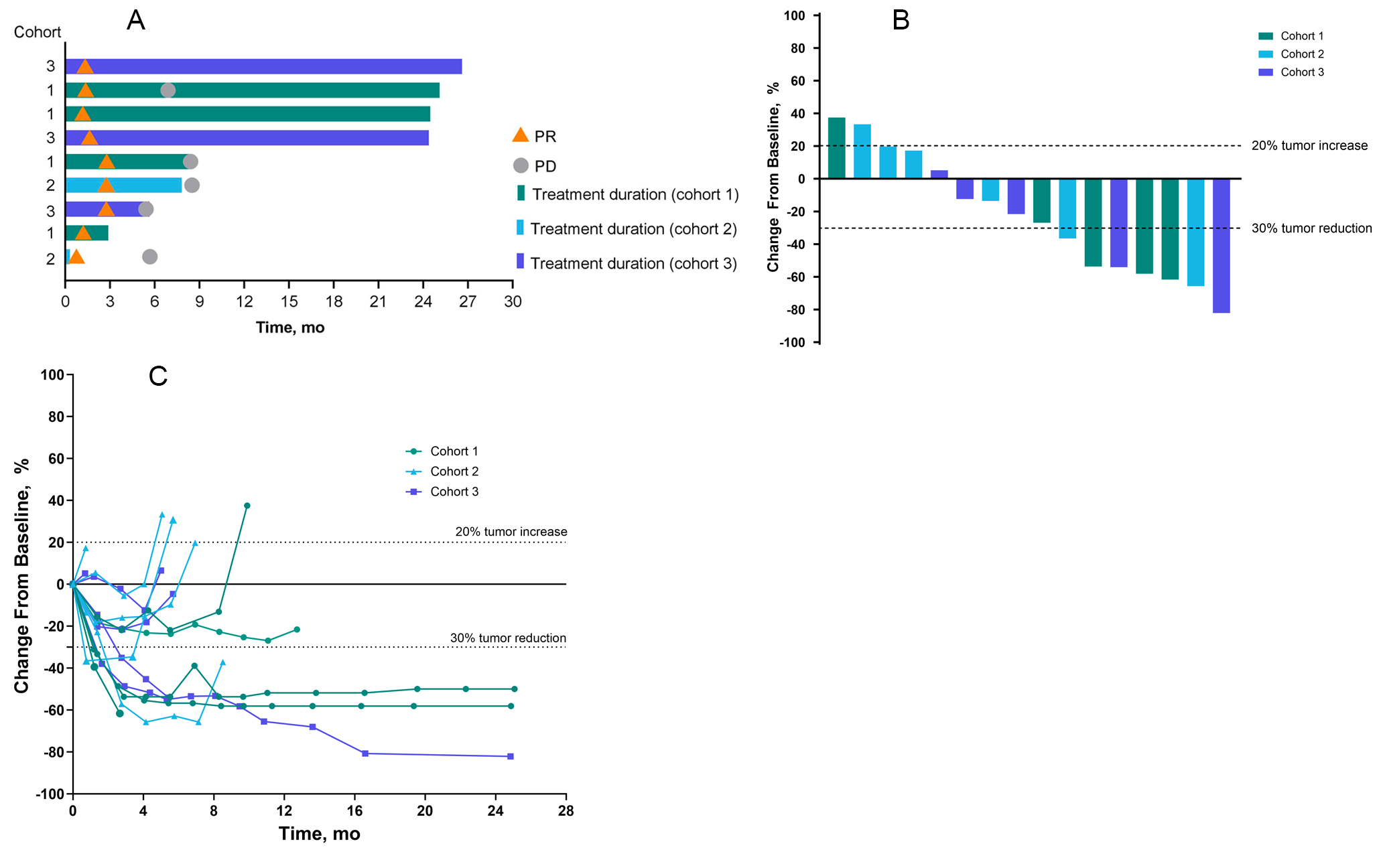
Best overall response (BOR) was determined using all time point responses (TPRs) up until the last evaluable TPR prior to or on the date of (i) radiological disease progression as defined by RECIST v1.1 or death; or (ii) loss to follow-up or withdrawal of consent; or (iii) receipt of subsequent anti-cancer medications, whichever was earlier. Investigator-assessed objective response rate (ORR) was defined as the proportion of patients with BOR of confirmed complete response (CR) or confirmed partial response (PR) according to RECIST v1.1. The interval for confirmation of CR and PR was at least 4 weeks. Disease control rate (DCR) was defined as the proportion of patients with a BOR of confirmed CR, confirmed PR, or stable disease (SD) lasting for at least 7 weeks. Duration of response (DoR) was calculated as the time (in months) from the date of first objective response (CR or PR confirmed after ≥ 4 weeks) until the date of the documented progression or death, whichever came first. PFS was defined as the time (months) from the date of first administration of study drug until the date of radiological disease progression or death due to any cause, whichever came first.
Statistical analysis
The safety analysis set consisted of patients who received at least 1 dose of fruquintinib and was used in safety and PFS analyses. The efficacy analysis set included patients who received at least 1 dose of fruquintinib, had measurable target lesions at baseline, and had either at least 1 tumor assessment after treatment, or no tumor assessment after treatment but clinical progression as noted by the Investigator, or death due to disease progression before their first tumor scan after treatment. ORR and DCR were calculated with 2-sided 95% confidence intervals (CIs) using the Clopper-Pearson method [7]. DoR and PFS were estimated using the Kaplan–Meier method [8]. DoR was analyzed for patients with confirmed CR or PR. All statistical analyses were performed using SAS® v9.4 or higher (SAS Institute, Cary, NC).



留言 (0)