
Remember me
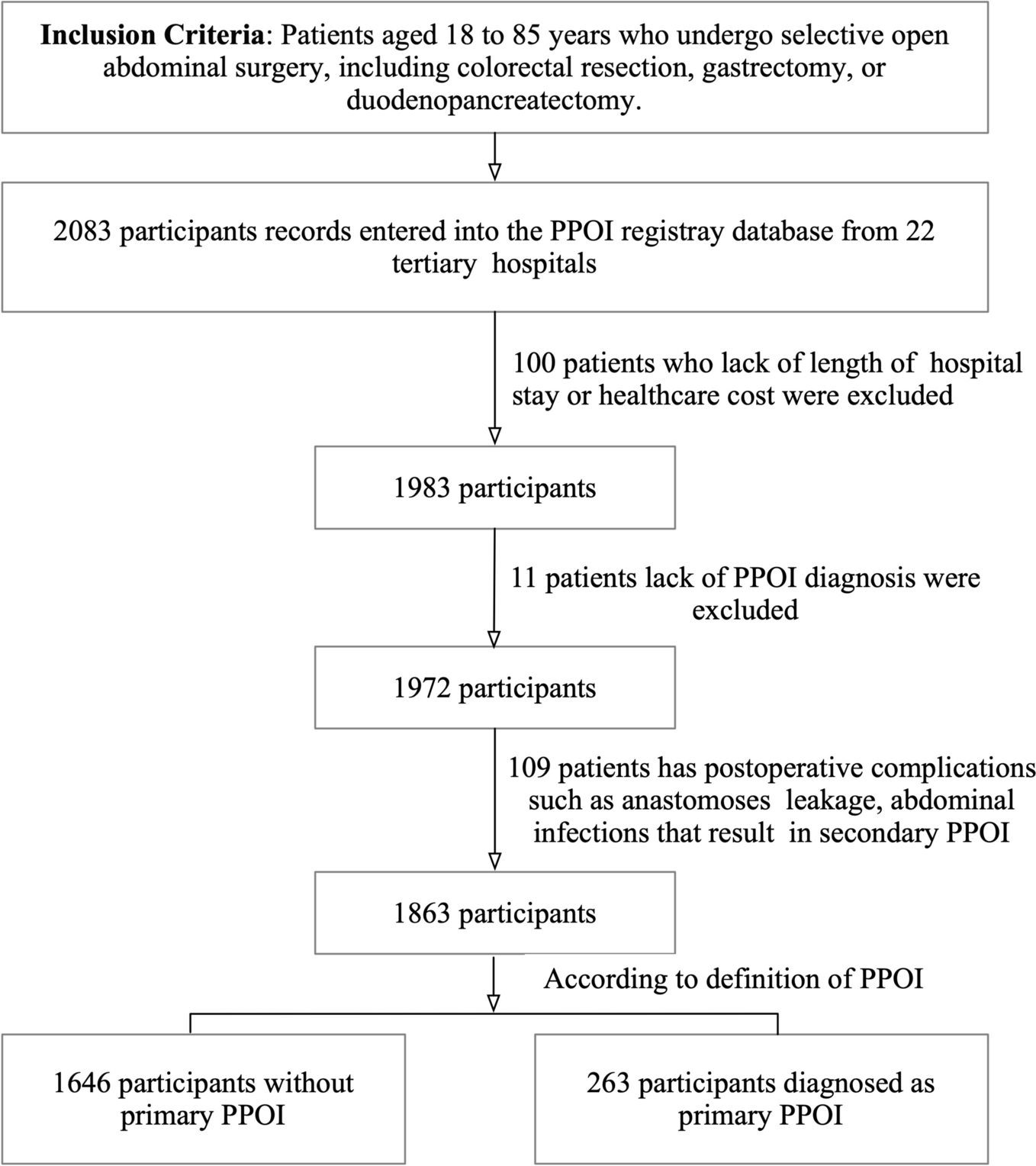
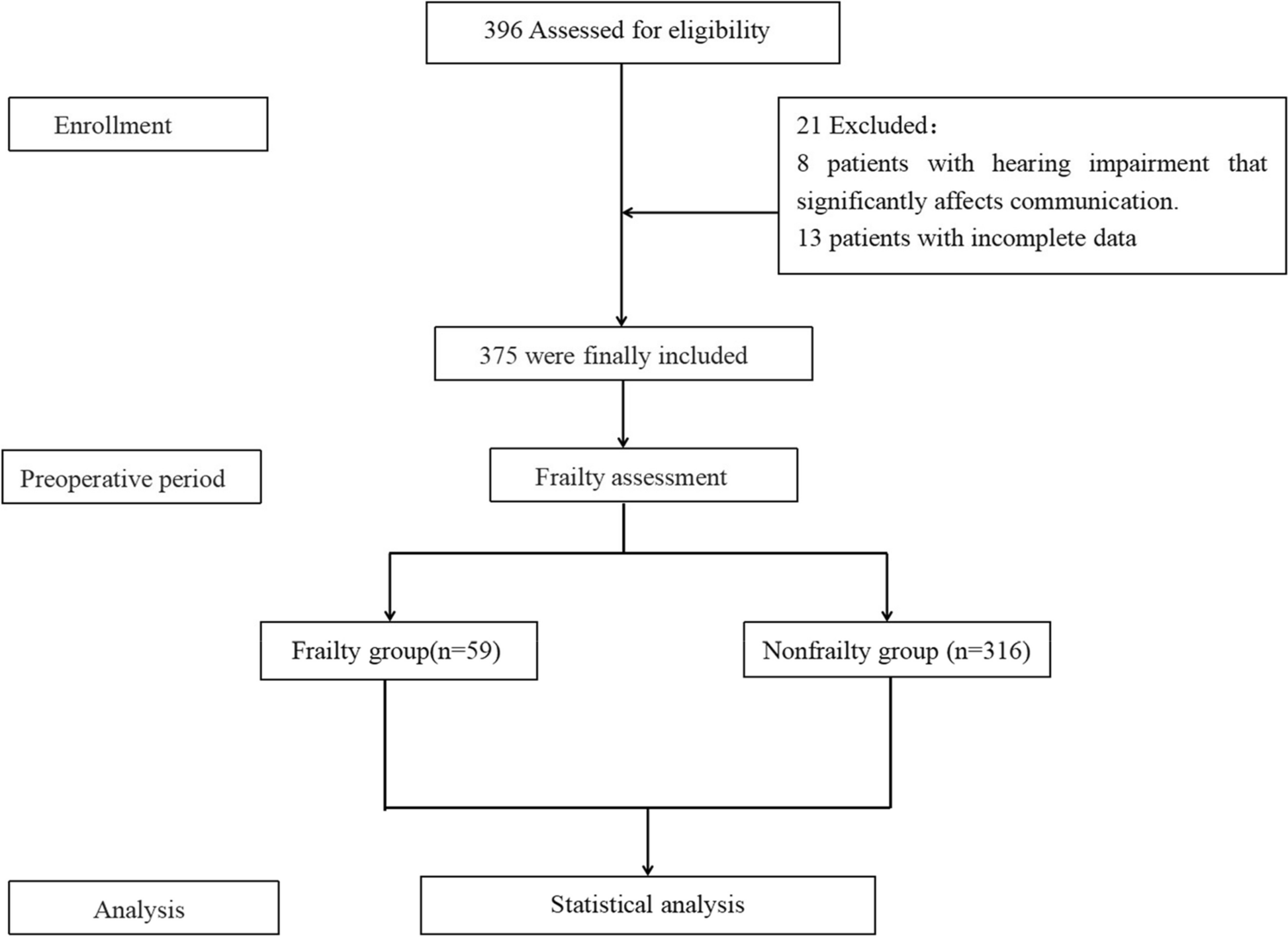
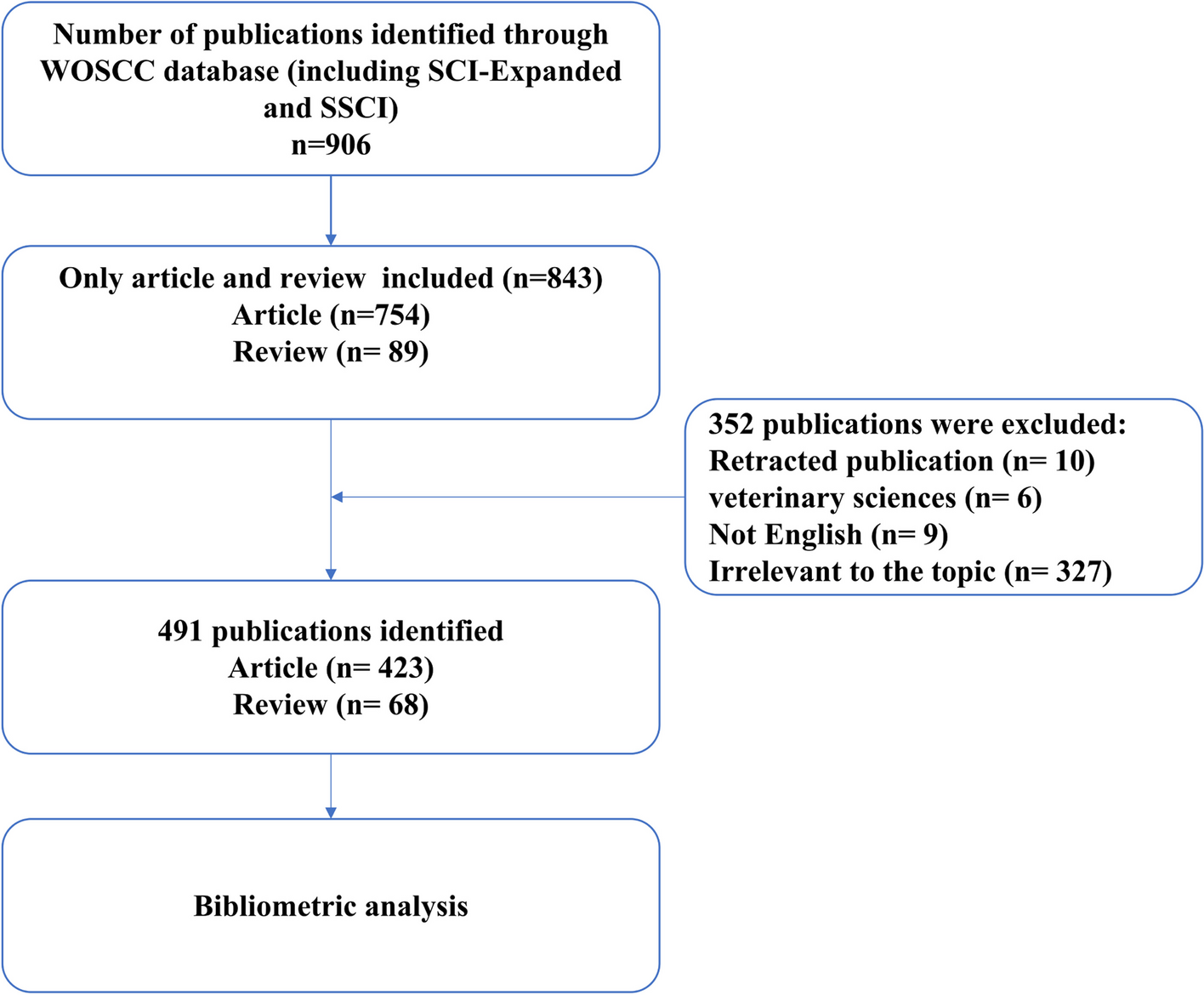
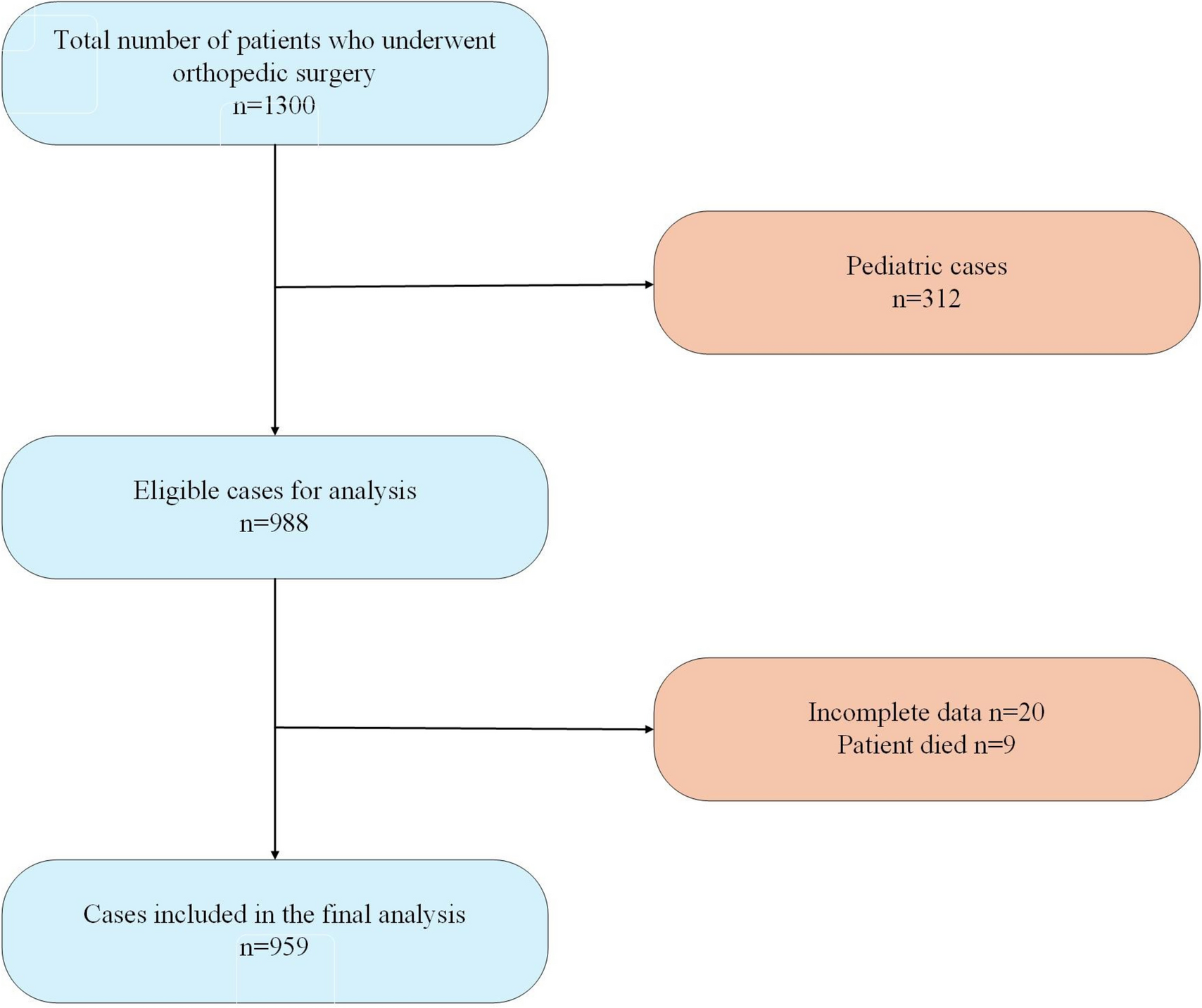


This study was approved by the local Institutional Review Board (IRB # 2021–00314) and written informed consent was obtained from all subjects participating in the trial. The trial was registered prior to patient enrollment at clinicaltrials.gov. The study was conducted between August 31, 2021, and January 19, 2022, when the last enrolled subject completed the final follow-up visit. The manuscript was prepared according to the applicable Consolidated Standards of Reporting Trials (CONSORT) guidelines (Gagnier et al. 2006).
Study participantsInclusion criteria were (1) adults at least 18 years of age up to 75 years of age, (2) able to provide informed written consent, and (3) willing to withhold use of chamomile products outside of the research study.
Exclusion criteria were (1) a past medical or family history of bleeding or thrombotic disorders; (2) taking chronic medications known to affect hemostasis, such as anticoagulants, antiplatelet agents, and selective-serotonin reuptake inhibitors; (3) a history of an abnormally elevated INR, prothrombin time (PT), activated partial thromboplastin time (aPTT), thrombin time (TT), or reptilase time (RT); (4) more than weekly non-steroidal anti-inflammatory drug use, e.g., aspirin, ibuprofen, or naproxen. (5) active intake of either ginger, garlic, ginkgo, ginseng, fish oil, black cohosh, feverfew, valerian, coenzyme q10, goldenseal, St. John Wort within 14 days of enrollment; (6) severe allergy to ragweed; (7) reported chamomile allergy; (8) consuming three or more alcoholic beverages daily; (9) active cigarette smoking; (10) female subjects who were pregnant, breast-feeding, or lactating; (11) hospitalization at time of screening; (12) scheduled surgical procedure during study period; (13) underweight subjects with body mass index < 18 kg/m2) or a history of malnourishment; (14) symptoms of active infection; (15) a history of estrogen-dependent condition such as uterine fibroids, breast cancer, uterine cancer, or ovarian cancer; (16) active intake of cyclosporine.
After informed written consent was obtained, baseline screening was completed and subjects were excluded (screen failed) if (1) the baseline complete blood count demonstrated a hematocrit below 30%, (2) platelet count below 150 × 103/μL, (3) white blood cell count below 3 × 103/μL or above 15 × 103/μL.
Objectives and hypothesisOur primary objective was to test the hypothesis that ingestion of chamomile prolongs the prothrombin time.
Study interventionsSummary of designThe study was a randomized, controlled cross-over design occurring over 5 weeks per subject (Fig. 2). After consent and screening tests, all subjects received the following three 7-day long interventions (3 chamomile teabags 3 times per day, 1 chamomile extract capsules 3 times per day, and 1 placebo capsules 3 times per day). Blood samples were obtained at the beginning and end of each weeklong intervention and sent to our hospital’s coagulation laboratory for analysis (see details below). There was a 1-week washout period in-between interventions. Subjects were randomized to the order in which the 3 interventions were received (see “Statistical methods” section and Fig. 2).
Fig. 2
Randomized order of interventions and laboratory sampling time points. Letters “A,” “B,” and “C” represent the three interventions (A = chamomile tea, B = chamomile capsule, and C = placebo capsule), therefore, there were 6 possible orders of interventions that subjects could be randomized to. A screening sample was collected within 72 h prior to initiation of the first study intervention (week 1). Subsequent blood samples were obtained before and after completing each 7-day (i.e., weeklong) intervention
Study products and chamomile preparations Chamomile tea drinkSubjects were instructed to consume a cup (approximately 5 oz.) of chamomile tea three times a day (morning, daytime, and evening). Tea was prepared using a widely available mass-produced tea bag (Celestial Seasonings®, USA). A total of three tea bags (1 g of dried flower in each tea bag) was used to make the cup of tea, which thus contained 3 g of tea. Therefore, over the course of the day subjects consumed 3 cups of tea, which contained a total tea dose of 9 g. The tea was steeped in either boiling water, water dispensed by an electric kettle that completes its heating cycle, or an on-demand heated water dispenser. Subjects were instructed that they should observe steam emerging from the cup once it is dispensed. Each dose of three tea bags (3 g total dose) was steeped in the 5 oz of hot water for 10 min. After the steeping period, tea bags were removed. Sugar and artificial sweeteners using aspartame, sucralose, or stevia extract were permitted as there is no established interaction between chamomile ingredients and sweeteners. Subjects were instructed to avoid adding other substances such as milk to the teacup during consumption. Subjects were also instructed to consume the tea shortly after meals, and to maintain their usual dietary habits. Subjects completed a daily log of the times they ingested the relevant tea and capsule (placebo or chamomile) as described below.
Chamomile powder extract in capsulesDuring a separate week (Fig. 2) subjects consumed chamomile extract capsules. The extract capsules were purchased from a single supplier, Swanson Superior Herbs- Chamomile Flower Extract-Standardized Apigenin 1.2% (Swanson Health Products, USA). Each of these capsules contains 500 mg of a chamomile extract which has been standardized by the manufacturer to 1.2% apigenin content by weight. A certificate of authenticity from a third-party analyst (Health Wright Products Inc., USA) was obtained from the capsule provider to ensure appropriate chamomile composition. To mask the content of the capsules (visually and odor), our hospital’s Investigational Pharmacy inserted the chamomile capsules into opaque blue capsules (Article No. 800223, Fagron Inc., USA). Subjects were instructed to consume one 500 mg capsule three times a day for a total daily dose of 1500 mg.
Placebo capsulesDuring a separate week (Fig. 2) subjects consumed a week of placebo capsules. Placebo capsules were prepared by our hospital’s Investigational Pharmacy using methylcellulose filler (Fagron Inc., USA) inserted into the same opaque blue container capsules used for the active drug and sealed to ensure blinding from compound appearance and smell.
Blood samplingVenous blood samples were collected by trained study staff, using sterile technique and a 21-gauge phlebotomy needle, at six time points throughout the study period (Fig. 2): (1) screening within 72 h prior to initiation of the first study intervention (day 0), (2) after completing the first 7-day intervention week (morning of day 8), (3) after 7 days washout of initial exposure, (4) after 7 days of second 7-day intervention, (5) after 7 days washout from second intervention, and (6) after 7 days of the third 7-day intervention.
At each of these 6 phlebotomy visits, 15 ml of blood were collected into citrated blood tubes (BD Vacutainer® Buff. Na Citrate 0.109 M, 3.2%, Becton, Dickinson and Company, USA). The blood tubes were delivered to our hospital’s Coagulation Clinical Laboratory within 2 h of sampling, which is earlier than the 4-h timeframe required by the laboratory. The blood samples were centrifuged in the laboratory to obtain platelet-poor plasma. Coagulation assays were completed on the platelet-poor plasma using the ACL TOP 750 CTS automatic coagulation analyzer (Werfen, Bedford, MA). Complete blood counts were performed on uncentrifuged blood using the Sysmex XN-10 hematology analyzer (Sysmex America, Lincolnshire, IL, USA).
Endpoints and statistical methodsData for this trial were entered into a dedicated REDCap database (REDCap, USA), which includes numerous safeguards to protect the integrity of the data. For example, logs are generated for all entry, revision, and deletion of data. All statistical analyses were performed by the study’s statistician (co-author JR).
The primary endpoint for the study was the change in PT from the baseline value at the start of each randomized 7-day long intervention. Secondary endpoints for the study included changes in the INR, aPTT, TT, RT, and FG from baseline values at the start of each randomized intervention. Safety endpoints included new easy bruising, new gum bleeding, or any new bleeding event occurring during the study. A separate standardized questionnaire including questions unrelated to coagulation or bleeding, e.g., sleep quality, was also obtained, and the results will be reported separately.
Randomization and designSubjects were randomized into a William’s square design, which is a special cross-over study design used to balance for carryover effects (Wang et al. 2009). Three 7-day long interventions (Fig. 2) were administered to each subject: (A) placebo, (B) chamomile extract capsule, and (C) chamomile tea, in varying order according to randomization assignment. A washout period lasting 7 days between treatments was employed to minimize any carryover effects. Since each subject received all three treatments, there were six possible sequences of treatment (ABC, ACB, BAC, BCA, CAB, CBA). Blocked randomization lists for sequence assignment were computed using SAS© 9.4 Software (SAS Institute Inc., USA). Each participant was randomly assigned one of the six treatment sequences using REDCap software, which provided allocation concealment. The study subjects, research coordinator (co-author D.H.) and the outcome assessor (co-author J.R.) were blinded from placebo versus chamomile extract capsule assignments by way of capsule covering, but they were not blinded from tea consumption.
Sample size and power calculationSample size calculations were performed using PASS Software (NCSS, LLC, USA) with a 3 × 3 cross-over design, Geisser-Greenhouse corrected F-test. The mean prothrombin time derived from a healthy reference sample at our institution was determined to be 11 s with a standard deviation of 0.7 s. We powered the study to detect a 10% change in our study groups, which we deemed as the smallest clinically relevant difference, approximately 1.1 s). Using a range of possible standard deviations, a range of correlations within a subject between 0.4 and 0.5, a power of 80%, and an alpha of 0.017 (adjusted for additional contrasts between treatment groups), the sample size required for the study was 12 participants. Therefore, each treatment sequence appeared twice within the study population.
Statistical analysesDemographic and medical history characteristics are described as frequency and percentages for categorical variables. With continuous variables, findings were reported using medians, interquartile ranges, or means and standard deviations (SD) as appropriate. Changes in the endpoints (measurement at completion of 7-day long intervention minus the baseline measurement immediately prior to starting the intervention) across the three study groups were analyzed using linear mixed models. Intervention type (tea, extract capsule, or placebo), randomization sequence, and period were treated as fixed effects, and subjects were treated as random effects. Carryover effects were examined using a period-intervention interaction term. This term was found to be insignificant and therefore removed from the model to reduce over-specification. Covariance structure specification was based on minimizing the Akaike’s Information Criterion. Adjusted contrasts were also examined between each pair of treatments. A p value of 0.05 was deemed significant for overall group comparisons, and an adjusted value of 0.017 was considered significant for paired contrasts between the groups. All calculations were performed using SAS© 9.4 Software (Cary, NC, USA).
Comments (0)