A fascinating method for predicting the major list mode of a ligand with a target protein with a known three-dimensional structure is molecular docking. It’s a crucial tool in the design of structure-based, computer-supported medicine. The intended benzimidazole derivations bind successfully with the target protein’s active site Voltage-gated Sodium Channel (NavMs)- 5HVX by using Autodock software. The designed emulsion A4, A6, A8 shows good list via hydrophobic and hydrogen bonds, respectively, to the target protein, whereas A1, A2, A3, A5, A7shows hydrophobic cling. The relations introduced by the active derivations within 5 Å compass to the list point of sodium channels and via a GABAergic pathway. In this exploration; all composites are active and A6 and A7 is the most active emulsion with minimal list affinity are named as potent impediments. Hydrophobic commerce of A8 and A7 with LEU168; LEU168; PHE142; LEU168; LEU138; MET175; PHE142; PHE172; LEU168; LEU138 a distinct group. The hydrogen bond conformation between the motes PHE and LEU is another factor is completely honored as indicated which have observed Tables 2 and 3. Docking investigations showed that the planned emulsion and target protein were the list mode of the most active composites. 2D and 3D Structure of designed benzimidazole derivations of A6; A7 and A8 are given (Figures 2, 3, 4).
Figure 1:
Designed benzimidazole derivatives
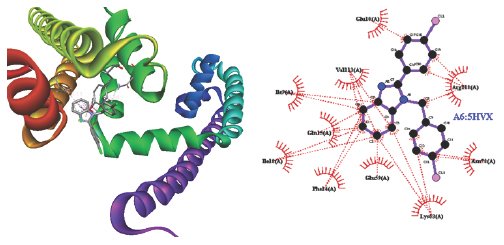
Figure 2:
2D and 3D Structure Designed Benzimidazole derivatives of A6
Comp.
Molecular weight (g/mol)
CMC rule violation
Lipinski’s rule violation
MolLog P
H bond donor
H bond acceptor
No. of Rotatable bonds
TPSA (Å2)
A1
432.86 g/mol
0
Yes
2.95
1
4
6
83.55 Å2
A2
412.44 g/mol
0
Yes
2.68
1
4
6
83.55 Å2
A3
443.41g/mol
0
Yes
2.40
1
6
7
129.37 Å2
A4
426.46 g/mol
0
Yes
2.88
1
4
7
83.55 Å2
A5
477.31 g/mol
0
Yes
3.05
1
4
6
83.55 Å2
A6
449.48 g/mol
0
Yes
3.15
1
4
6
83.55 Å2
A7
428.44g/mol
0
Yes
2.15
1
5
7
92.78 Å2
A8
398.41g/mol
0
Yes
2.47
1
4
6
83.55 Å2
A9
416.40g/mol
0
Yes
2.84
1
5
6
83.55 Å2
A10
423.42 g/mol
0
Yes
1.81
1
5
6
107.34 Å2
Table 2:
Druglikeness analysis of derivatives of designed compound of benzimidazole
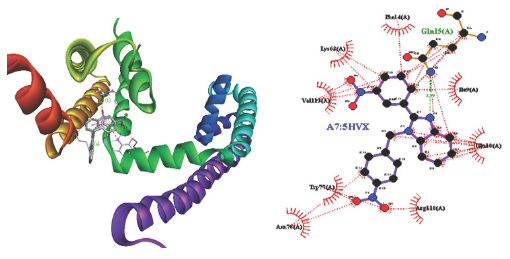
Figure 3:
2 D and 3 D Structure Designed Benzimidazole derivatives of A7
Active Amino acid
Bond length
Bond Type
Bond Category
Ligand Energy
Docking score
A1
LEU168
3.36354
Hydrogen Bond
Carbon Hydrogen Bond
22.9420 kcal/mol
-8.1
LEU168
3.55906
Hydrophobic
Pi-Sigma
LEU168
3.86165
Hydrophobic
Pi-Sigma
PHE142
3.80812
Hydrophobic
Pi-Pi Stacked
PHE172
4.06452
Hydrophobic
Pi-Pi Stacked
LEU138
5.32262
Hydrophobic
Pi-Alkyl
A2
THR139
2.49397
Hydrogen Bond
ConventionalHydrogen Bond
34.4955 kcal/mol
-7.8
LEU168
3.42119
Hydrophobic
Pi-Sigma
PHE172
4.16443
Hydrophobic
Pi-Pi Stacked
PHE172
4.26798
Hydrophobic
Pi-Pi Stacked
PHE142
4.96145
Hydrophobic
Pi-Pi T-shaped
ALA135
4.3971
Hydrophobic
Alkyl
LEU138
4.32799
Hydrophobic
Alkyl
TYR143
5.25675
Hydrophobic
Pi-Alkyl
LEU138
5.3909
Hydrophobic
Pi-Alkyl
A3
LEU168
3.79525
Hydrogen Bond
Carbon Hydrogen Bond
21.0582 kcal/mol
-8.3
THR139
3.50541
Hydrogen Bond
Carbon Hydrogen Bond
LEU168
3.81015
Hydrophobic
Pi-Sigma
LEU168
3.58694
Hydrophobic
Pi-Sigma
PHE172
4.16592
Hydrophobic
Pi-Pi Stacked
PHE172
4.2107
Hydrophobic
Pi-Pi Stacked
UNL1
4.87253
Hydrophobic
Pi-Pi Stacked
PHE142
4.77548
Hydrophobic
Pi-Pi T-shaped
LEU168
4.94469
Hydrophobic
Alkyl
LEU138
5.35849
Hydrophobic
Pi-Alkyl
A4
THR139
2.32321
Hydrogen Bond
ConventionalHydrogen Bond
20.5745 kcal/mol
-7.9
LEU168
3.5664
Hydrophobic
Pi-Sigma
PHE172
4.09765
Hydrophobic
Pi-Pi Stacked
PHE172
4.33962
Hydrophobic
Pi-Pi Stacked
UNL1
4.26071
Hydrophobic
Pi-Pi Stacked
PHE142
4.83269
Hydrophobic
Pi-Pi T-shaped
LEU138
5.26085
Hydrophobic
Pi-Alkyl
A5
THR139
2.27351
Hydrogen Bond
ConventionalHydrogen Bond
15.7829 kcal/mol
-8.5
LEU168
3.76897
Hydrogen Bond
Carbon Hydrogen Bond
LEU168
3.48472
Hydrophobic
Pi-Sigma
PHE142
5.76341
Hydrophobic
Pi-Pi Stacked
PHE172
4.31901
Hydrophobic
Pi-Pi Stacked
PHE172
4.18386
Hydrophobic
Pi-Pi Stacked
PHE142
5.27218
Hydrophobic
Pi-Pi T-shaped
A6
LEU168
3.94059
Hydrophobic
Pi-Sigma
23.7210 kcal/mol
-8.1
LEU168
3.58577
Hydrophobic
Pi-Sigma
PHE142
3.75521
Hydrophobic
Pi-Pi Stacked
LEU168
5.38106
Hydrophobic
Alkyl
LEU138
4.31334
Hydrophobic
Alkyl
MET175
4.66181
Hydrophobic
Alkyl
PHE142
4.45107
Hydrophobic
Pi-Alkyl
PHE172
5.21336
Hydrophobic
Pi-Alkyl
LEU168
4.04581
Hydrophobic
Pi-Alkyl
LEU138
5.41196
Hydrophobic
Pi-Alkyl
A7
THR139
2.27153
Hydrogen Bond
ConventionalHydrogen Bond
26.7202 kcal/mol
-8.7
LEU168
3.7803
Hydrogen Bond
Carbon Hydrogen Bond
THR139
3.48539
Hydrogen Bond
Carbon Hydrogen Bond
ALA135
3.35577
Hydrogen Bond
Carbon Hydrogen Bond
UNL1
3.72372
Hydrogen Bond
Carbon Hydrogen Bond
LEU168
3.81841
Hydrophobic
Pi-Sigma
LEU168
3.59081
Hydrophobic
Pi-Sigma
PHE172
4.22858
Hydrophobic
Pi-Pi Stacked
PHE172
4.19296
Hydrophobic
Pi-Pi Stacked
UNL1
4.84604
Hydrophobic
Pi-Pi Stacked
LEU168
4.96597
Hydrophobic
Alkyl
LEU168
4.92852
Hydrophobic
Alkyl
LEU138
4.06661
Hydrophobic
Alkyl
A8
THR139
2.30219
Hydrogen Bond
ConventionalHydrogen Bond
44.7859 kcal/mol
-7.8
THR13
3.5655
Hydrogen Bond
Carbon Hydrogen Bond
ALA135
3.34208
Hydrogen Bond
Carbon Hydrogen Bond
LEU168
3.80109
Hydrophobic
Pi-Sigma
LEU168
3.60123
Hydrophobic
Pi-Sigma
PHE172
4.25657
Hydrophobic
Pi-Pi Stacked
PHE172
4.21428
Hydrophobic
Pi-Pi Stacked
UNL1
4.90046
Hydrophobic
Pi-Pi Stacked
LEU168
4.98171
Hydrophobic
Alkyl
LEU168
4.89463
Hydrophobic
Alkyl
LEU138
4.06486
Hydrophobic
Alkyl
Table 3:
The active amino residues, bond length, bond category, bond type, ligand energies, and docking scores of benzimidazole

Figure 4:
2D and 3D Structure Designed Benzimidazole derivatives of A8

Comments (0)