
記住我




The efficacy and safety of antidepressants (ADs) in bipolar disorder (BD) remain controversial because of the lack of a consistent evidence base and potential concern for mood destabilization.1 This controversy has only been magnified with recent data reporting a doubling (up to 40%), over the course of 20 years, of AD monotherapy prescriptions for BD.2 Given the predominance of bipolar depression and its underdeveloped pharmacopeia, clinical research is needed to identify predictors of AD nonresponse or adverse events. A risk mitigation model-based approach to AD use can impact clinical practice by decreasing exposure to unsuccessful treatment trials or drug-associated adverse events. Exemplary of the lack of precision in AD therapy in major depression are data from the Sequenced Treatment Alternatives to Relieve Depression study, which reported an overall remission rate of 67% requiring up to 4 AD treatment trials over the course of 56 weeks.3,4
Several operationalized terms have invariably been used to describe the clinical phenomena of mood destabilization such as treatment emergent or AD-induced mania5 treatment emergent affective switch,6 chronic irritable dysphoria,7 and cycle acceleration.8 Prevalence and incidence rates for treatment emergent mania (TEM) have varied across studies mainly because of methodological differences in study design and cohort characteristics. General incidence rate estimates in retrospective studies are up to 30%, which are generally greater than prospective studies such as Embolden II where TEM for paroxetine monotherapy (11%) was 3 times higher than quetiapine (2%–4%).5,9 In the latest clinical review, 5 disease characteristics were associated with TEM, including female sex, early age of onset, BD-I subtype (vs II), and greater number of prior episodes (total depressive episodes).10 Moreover, this meta-analysis found an association only with prior number of depressive episodes and increased risk for TEM (P = 0.008).10
One of the most well-studied genetic variants in TEM is a repeat polymorphism located in the promoter region of the serotonin transporter gene (5-HTTLPR; SLC6A4), specifically, its long (l) and short (s) allele variants, which lead to differences (high vs low, respectively) in transcriptional activity of the transporter protein. Previous studies have suggested that the l/l genotype, in comparison to s/s, had higher response rates largely in Caucasians11; in contrast, better treatment response in patients of East Asian ethnicity was associated with the s/s genotype.12 To date, research suggests a trend (P = 0.057) association between the s allele of the 5-HTTLPR polymorphism of SLC6A4 and TEM.5,13 The purpose of this study was to review the association of the 5-HTTLPR polymorphism of SLC6A4 with TEM in BD by providing an updated meta-analysis alongside starting a discussion on the merits of developing risk stratification models to guide when not to provide AD treatment for bipolar depression.
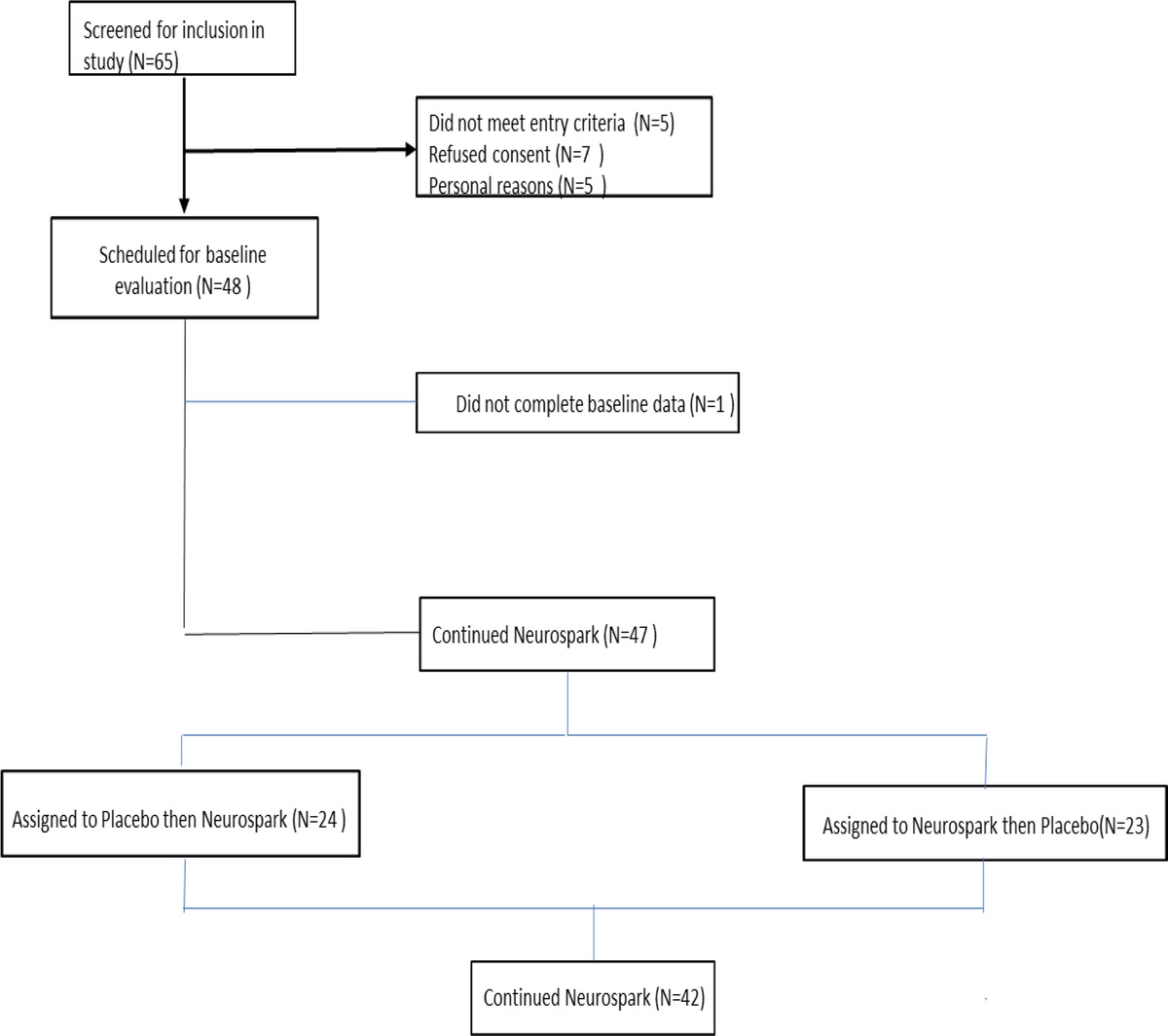
METHODSA total of 142 articles were screened, yielding a total of 76 studies. After review using the Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines,14 7 studies referencing the 5-HTTLPR polymorphism of SLC6A4 and TEM were included in the meta-analysis (Fig. 1). Studies were composed of both adolescent and adult populations, whereas our initial meta-analysis, limiting age as a potential confounding variable, included only studies in adults.5 The effect size was calculated using odds ratio under the Der-Simonian and Laird random effects model with an α level of 0.05 chosen for statistical significance. This work was performed in R (v3.6.1)15 using the “meta” (v4.9.6, http://cran.r-project.org/web/packages/rmeta/index.html) package.16 The list of search terms used and how they are combined as well as study inclusion/exclusion criteria can be found in Table S1 and S2 of the Supplemental Digital Content (https://links.lww.com/JCP/A873). The assessment of methodological quality of the randomized controlled trials included in the meta-analysis was done using the Cochrane Collaboration's risk of bias tool assessing 6 domains: sequence generation, allocation concealment, blinding of study participants and personnel, blinding of the outcome assessment, selective reporting, incomplete outcome data, and other biases.17
 FIGURE 1:
FIGURE 1: Flow diagram of the study procedure for study selection.
For this study, we initially reviewed and modified the International Society of Bipolar Disorders nomenclature of treatment emergent affective switch. The International Society of Bipolar Disorders definition globally considers timing (up to 12 weeks), duration, and severity of mood symptoms in relation to the intervention.6 This study focused only on the manic pole of the illness and its association with the use of ADs, with a narrower window of exposure. Therefore, we used the term AD-associated TEM throughout the manuscript.
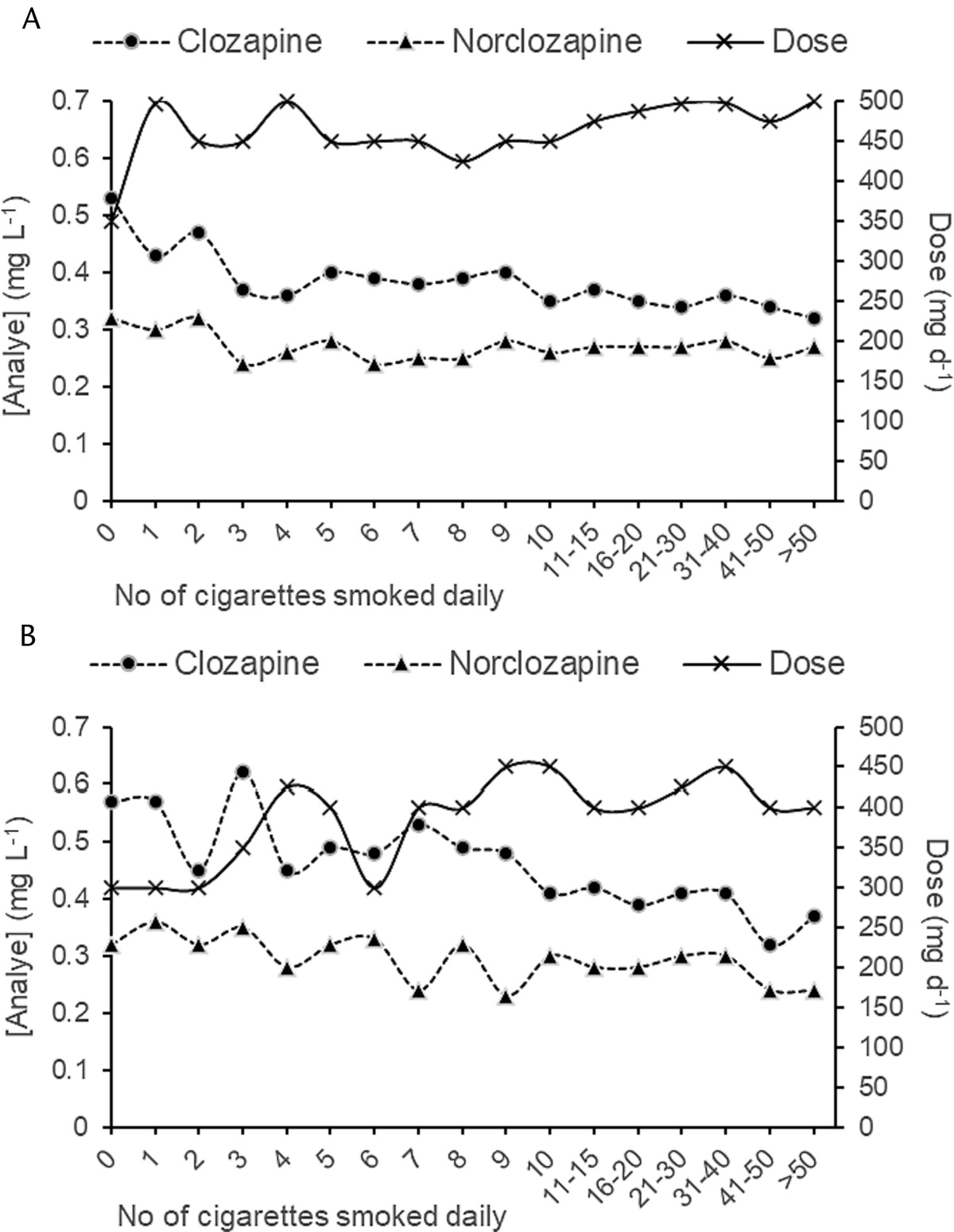
RESULTSSeven studies were included in the meta-analysis (N = 1578, TEM = 594, No-TEM = 984)5,18–23 with a total mean age of 35.0 ± 10.89 years and 41% of the sample being men. Table 1 summarizes definitions of TEM and any methods used to control for confounding variables. Only 4 of the 7 studies5,18,22,23 reported a time duration between the start of the AD to the emergence of TEM ranging from 4 to 12 weeks. Most studies included multiple proserotoninergic ADs and concurrent mood stabilizer treatment. Only 2 studies included patients on AD monotherapy,18,23 one of which22,23 examined the association of TEM and potential clinical risk factors, such as a rapid cycling phenotype. Quality assessment of the included studies revealed an overall low risk of bias. Our meta-analysis using a random effects model identified a nominally significant association between the s allele of the 5-HTTLPR polymorphism and TEM in BD (odds ratio, 1.434; 95% confidence interval, 1.001–2.055; P = 0.0493; I2 = 52%) (Fig. 2).
TABLE 1 - Description of Included Studies TEM Phenotype Definitions Study TEM Phenotype Definition Mundo et al (2001)21 DSM-IV diagnostic confirmation of BD-I/BD-II and DSM-IV criteria for mania-hypomania. No time duration for drug exposure. Multiple proserotoninergic AD. Rousseva et al (2003)22 Diagnostic Interview for Genetic Studies for diagnosis of BD-I, BD-II, or BD-NOS. Used both a broad TEM definition by self-reports and time duration within 12 wk of starting AD. Multiple AD classes. Serretti et al (2004)18 DSM diagnostic confirmation BD-I or BD-II and mania/hypomania fulfilling DSM-IV criteria within 4 wk from starting AD. Multiple AD classes. Baumer et al (2006)20 Pediatric population. WASH-U-KSADS confirmation of BD-I or BD-II. Two definitions: A, 1 d of expansive euphoric irritable mood plus 3 other cardinal symptoms fulfilling DSM-IV criteria. B, increase of more than 4 points on the YMRS behavioral scale for 12 wk of treatment or a dose increase. Mostly used SSRIs. Family history of parent with BD. Masoliver et al (2006)19 DSM-IV diagnostic confirmation of BD-I/BD-II or BD-NOS. No duration time frame provided. de Aguiar Ferreira et al (2009)23 DSM-IV diagnostic confirmation of BD-I/BD-II or BD-NOS. Time duration of episode within the 8 wk of treatment. Frye et al (2015)5 DSM diagnostic confirmation BD-I/BD-II; manic episode by DSM criteria during an 8-wk time frame from starting an AD or increasing AD dose.BD-NOS indicates BD not specified; DSM-IV, Diagnostic and Statistical Manual of Mental Disorders, 4th Edition; SSRI, selective serotonin reuptake inhibitor; WASH-U-KSADS, Washington University Kiddie Schedule for Affective Disorders and Schizophrenia; YMRS, Young Mania Rating Scale.
 FIGURE 2:
FIGURE 2: SLC6A4 s allele and TEM.
CONCLUSIONThis meta-analysis identified an association between the s allele of the 5-HTTLPR polymorphism with TEM in BD. Although this association is nominal, it is the first nonclinical marker that has a significant relationship with an AD adverse event, suggesting it may be a potential biomarker. The serotonin transporter variant 5-HTTLPR is widely available in many commercial decision support tools. Although these results are potentially quite useful for clinical practice, these commercial platforms have largely focused on nonbipolar clinical patients. In comparison to our previous work,13 this nominal significance is based on adding earlier work from an adolescent cohort by Baumer et al.20 Furthermore, age10 has been identified as a risk factor for TEM and argues for future risk models to incorporate known clinical and nonclinical risk factors for TEM.
Although a nominal association was identified with the serotonin transporter and TEM, what is also more relevant is the absence of studies that have examined the contribution of norepinephrine (NET, SLC6A2) or dopamine transporters (DAT1, SLC6A3)/receptors (DRD2, DRD4) in TEM. Although the serotonin transporter genetic variation is commercially available in many pharmacogenomic decision support tools, greater efforts, more broadly, should focus on examining pharmacokinetic (PK)-pharmacodynamic (PD) interactions and genome-wide approaches to determine genetic variants that may contribute to TEM.
Moreover, most of the previous literature has focused on single pharmacogenomic variants and treatment response with only a recent shift to study PK-PD interaction. For example, a candidate gene study identified nominal association between a norepinephrine transporter gene (NET; SLC6A2) variant and response to nortriptyline.24 A second study has suggested an association between NET; SLC6A2 and SERT; SLC6A4 and treatment remission with venlafaxine.25 More recently, PK (CYP2D6, CYP2C19) and PD (SLC6A4, SLC6A2) genetic variations were investigated in patients with major depressive disorder (MDD) treated with an serotonin norepinephrine reuptake inhibitor agent who had previously failed prospective treatment with an SSRI. There was a significant interaction in patients with the SLC6A4 l/l genotype and CYP2D6 ultra-rapid phenotype being significantly more likely to achieve remission with venlafaxine XR in comparison to those expressing an intermediate or poor metabolizer phenotype.26 Considering our PD correlates of TEM and uncontrolled observations of PK correlates of TEM in bipolar patients who were poor/intermediate metabolizers at CYP2D6 coprescribed 2D6 substrate and inhibitor of CYP2D6 (ie, fluoxetine, paroxetine),27 future PK-PD studies targeting antidepressants in BD are encouraged.
Molecular and pharmacogenomic studies conducted in MDD patients can inform future studies and modeling of ADs in BD. For example, the mammalian target of rapamycin and the Wnt signaling pathway have gained considerable attention due that mechanistically they are involved in neurogenesis, protein synthesis, and synaptic plasticity; these pathways have been identified as possibly contributing to AD response, although these warrant further investigation in AD studies in both MDD and BD.28 Second, mitochondrial energetics are increasingly implicated in the underlying neurobiology of BD.29 There is growing evidence of a possible association between mitochondrial dysfunction and alterations in oxidative phosphorylation, inflammation, and neuronal excitotoxicity,30–32 which may contribute to perpetuation of the illness.33
There are limitations to our meta-analysis. First, we should consider this as a small meta-analysis. According to the Cochrane Library (n = 7 studies), this small study limits the ability to examine publication bias and account for the I2-reported heterogeneity.34,35 The heterogeneity (I2 = 52%) may, in part, be related to the lack of addressing confounding variables in some studies and a universal standardized definition of TEM. Nonetheless, the time from the start of AD to TEM aligns with the window of vulnerability from a large Swedish registry (N = 3200), which reported a higher risk of TEM in adult BD-I patients treated with AD monotherapy, with the greatest risk occurring during the first 12 weeks of treatment (hazard ratio = 2.83; 95% confidence interval, 1.12–7.19).36 This risk period of short-term AD use may involve differences in molecular signaling and/or upregulation of mitochondrial pathways37 compared with chronic AD exposure.38 Another potential source of heterogeneity was not controlling for age of BD onset and the total number of prior episodes.10 For example, Mundo et al18,39 included a population of BD patients with a younger earlier mean age of BD onset (approximately 19.8 years) compared with Serretti et al (approximately 32 years of age). In addition, inherent to limitations of the meta-analysis, our overall quantitative summary effect may not account for important differences between studies, and we could not disregard the account for a false-positive rate by the use of the Der-Simonian-Laird method. Lastly, we should consider that our analysis is focused on only one genetic variant instead of an overall comparison of multiple variants.
Nevertheless, our preliminary data may begin to pave an initial roadmap for the development of risk models that could potentially include different biological measures alongside known clinical risk factors (ie, BD-I subtype, female gender, number of depressive episodes, absence of concurrent antimanic mood stabilizer, and type of AD medication [serotonin norepinephrine reuptake inhibitor vs SSRI]) to create a composite risk score for TEM. Actionable pharmacogenomic variants could include a cytochrome P450 (CYP) metabolizer phenotype, variation in monoamine transporters such as SLC6A4 and SLC6A2, their interaction, and any additional targets identified in previous genome wide association studies. Integrating omics and large clinical datasets and employing machine learning (ML) algorithms may provide additional risk stratification tools for clinicians individualizing treatment recommendations. For example, there is precedence in using artificial intelligence to predict both treatment resistance and response in MDD as well as lithium response in BD.40–42 The possible advantages of ML in predicting personalized treatment care and emergence of adverse events already have been developed in other fields of medicine. For example, Bitencourt et al43 incorporated clinical information and magnetic resonance imaging features from breast cancer patients to develop a model with 83.9% accuracy in predicting response to neoadjuvant chemotherapy. Similarly, Farahmand et al44 proposed a classifier model, which achieved up to 90% of accuracy in predicting HER2 status and trastuzumab response in breast cancer patients. Finally, studies incorporating multiomics through data integration and ML have also contributed to determine response to therapy.45
Therefore, ML models may have the potential to include large data (clinical and genetic) from electronic health records to stratify and refine patients' characteristics into different categories of risk for TEM. These models could estimate a probability of 3 states: “no risk, low risk, or high risk.” Importantly, the development of advanced algorithms from ML could aid in phenotyping patients from different electronic health records and evaluate its performance in prediction of TEM. Thus, the performance of the prediction model could be estimated by the area under the receiver operating curve by comparing its results to a criterion standard (identified TEM case by clinician). Moreover, these TEM risk phenotypes could serve the purpose for identification of risk and to better understand the intricacies of the TEM phenotype. A potential type of model may incorporate information extracted from medical records, including demographics, clinical history, AD type and dosage, comorbid medication use as well as information on an individual's genetic variation, including CYP metabolizer phenotypes and other variants discovered in genome wide association studies of AD treatment response/adverse outcomes. For example, a potential model configuration would rank a 20-year-old woman struggling with depression with a history of mania (but not currently on a mood stabilizer), family history of BD-I, a CYP2D6 poor metabolizer phenotype, and an SLC6A4 reduced expression (s allele) phenotype as high risk, whereas a 50-year-old man with treatment resistant depression, with older age of onset, already prescribed atypical antipsychotic augmentation, with a CYP2D6 extensive (normal) metabolizer phenotype, and with an SLC6A4 normal expression (l allele) phenotype as low risk. Importantly, these models would require repeated cross-validation and out-of-sample validation to ensure an optimized model with high accuracy, sensitivity, and specificity with minimal bias.46,47 Subsequent models would benefit from ongoing updates and more data as new risk factors may be identified. Although further large-scale longitudinal studies will be needed to develop and test the clinical utility of these potential risk stratification models for TEM, these models could transform personalized medicine practice in BD by providing evidence-guided recommendations and improving patients' outcomes.
ACKNOWLEDGMENTSFunding for the study was provided by the Marriott Foundation and the Dauton Family. The foundation and family had no further role in the study design, analysis or interpretation of the data, in the writing of the report, or in the decision to submit the article for publication. We would like to thank Mr Larry Prokop from the Mayo Clinic Library, Mayo Clinic Rochester, MN, for helping and conducting the literature search.
AUTHOR DISCLOSURE INFORMATIONConflicts of Interest: N. A. N., B. J. C., L. M. B., M. G. R., M. V., A. O., J. S. R., A. C. B., J. G. L., and J. M. B. report no financial relationships with commercial interests.
B. S. received grant support from Mayo Clinic.
F. R. N. receives grant support from the National Institute of Mental Health K23 Award (K23MH120503) and from a 2017 NARSAD Young Investigator Award from the Brain and Behavior Research Foundation; has a US Patent and Trademark Office patent # 10,857,356; receives consultant fees from Otsuka Pharmaceutical; and has received nonfinancial research support from Soterix Medical.
M. L. P. has served on an advisory board of Janssen and receives grant support from ANID FONDECYT Regular 1181365 and FONDEF ID19I10116.
S. L. M. is or has been a consultant to or member of the scientific advisory boards of Avanir, Allergan (now AbbVie), Bracket (now Signant Health), Naurex, Idorsia, Intra-Cellular Therapies, Inc, Shire (now Takeda), Sunovion, and Takeda. She is or has been a principal or coinvestigator on studies sponsored by the Agency for Healthcare Research and Quality, Avenir, AstraZeneca, Cephalon, Forest, Marriott Foundation, Medibio, National Institute of Mental Health, Orexigen Therapeutics, Inc, Jazz, Shire (now Takeda), Sunovian, and Takeda Pharmaceutical Company Ltd. She is also an inventor on United States Patent No. 6,323,236 B2, Use of Sulfamate Derivatives for Treating Impulse Control Disorders, and along with the patent's assignee, University of Cincinnati, Cincinnati, OH, has received payments from Johnson & Johnson, which has exclusive rights under the patent.
M. A. F. has received grant support from Assurex Health and Dauton Family, currently from Mayo Foundation, received CME travel and honoraria from Carnot Laboratories, and has financial interest/stock ownership/royalties from Chymia LLC.
REFERENCES 1. Pacchiarotti I, Bond DJ, Baldessarini RJ, et al. The International Society for Bipolar Disorders (ISBD) task force report on antidepressant use in bipolar disorders. Am J Psychiatry. 2013;170:1249–1262. 2. Rhee TG, Olfson M, Nierenberg AA, et al. 20-year trends in the pharmacologic treatment of bipolar disorder by psychiatrists in outpatient care settings. Am J Psychiatry. 2020;177:706–715. 3. Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med. 2006;354:1243–1252. 4. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–1917. 5. Frye MA, McElroy SL, Prieto ML, et al. Clinical risk factors and serotonin transporter gene variants associated with antidepressant-induced mania. J Clin Psychiatry. 2015;76:174–180. 6. Tohen M, Frank E, Bowden CL, et al. The International Society for Bipolar Disorders (ISBD) task force report on the nomenclature of course and outcome in bipolar disorders. Bipolar Disord. 2009;11:453–473. 7. El-Mallakh RS, Ghaemi SN, Sagduyu K, et al. Antidepressant-associated chronic irritable dysphoria (ACID) in STEP-BD patients. J Affect Disord. 2008;111(2–3):372–377. 8. Altshuler LL, Post RM, Leverich GS, et al. Antidepressant-induced mania and cycle acceleration: a controversy revisited. Am J Psychiatry. 1995;152:1130–1138. 9. McElroy SL, Weisler RH, Chang W, et al. A double-blind, placebo-controlled study of quetiapine and paroxetine as monotherapy in adults with bipolar depression (EMBOLDEN II). J Clin Psychiatry. 2010;71:163–174. 10. Melhuish Beaupre LM, Tiwari AK, Gonçalves VF, et al. Antidepressant-associated mania in bipolar disorder: a review and meta-analysis of potential clinical and genetic risk factors. J Clin Psychopharmacol. 2020;40:180–185. 11. Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry. 2010;15:473–500. 12. Ng CH, Easteal S, Tan S, et al. Serotonin transporter polymorphisms and clinical response to sertraline across ethnicities. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:953–957. 13. Biernacka JM, McElroy SL, Crow S, et al. Pharmacogenomics of antidepressant induced mania: a review and meta-analysis of the serotonin transporter gene (5HTTLPR) association. J Affect Disord. 2012;136(1–2):e21–e29. 14. Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. Int J Surg. 2021;88:105906. 15. R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2022. Available at: http://wwwR-projectorg/: R Foundation for Statistical Computing. Accessed December 20, 2022. 16. Balduzzi S, Rücker G, Schwarzer G. How to perform a meta-analysis with R: a practical tutorial. Evid Based Ment Health. 2019;22:153–160. 17. Higgins JP, Altman DG, Gøtzsche PC, et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ. 2011;343:d5928. 18. Serretti A, Artioli P, Zanardi R, et al. Genetic features of antidepressant induced mania and hypo-mania in bipolar disorder. Psychopharmacology (Berl). 2004;174:504–511. 19. Masoliver E, Menoyo A, Pérez V, et al. Serotonin transporter linked promoter (polymorphism) in the serotonin transporter gene may be associated with antidepressant-induced mania in bipolar disorder. Psychiatr Genet. 2006;16:25–29. 20. Baumer FM, Howe M, Gallelli K, et al. A pilot study of antidepressant-induced mania in pediatric bipolar disorder: characteristics, risk factors, and the serotonin transporter gene. Biol Psychiatry. 2006;60:1005–1012. 21. Mundo E, Walker M, Cate T, et al. The role of serotonin transporter protein gene in antidepressant-induced mania in bipolar disorder: preliminary findings. Arch Gen Psychiatry. 2001;58:539–544. 22. Rousseva A, Henry C, Van Den Bulke D, et al. Antidepressant-induced mania, rapid cycling and the serotonin transporter gene polymorphism. Pharmacogenomics J. 2003;3:101–104. 23. de Aguiar Ferreira A, Neves FS, da Rocha FF, et al. The role of 5-HTTLPR polymorphism in antidepressant-associated mania in bipolar disorder. J Affect Disord 2009;112(1–3):267–272. 24. Uher R, Huezo-Diaz P, Perroud N, et al. Genetic predictors of response to antidepressants in the GENDEP project. Pharmacogenomics J. 2009;9:225–233. 25. Marshe VS, Maciukiewicz M, Rej S, et al. Norepinephrine transporter gene variants and remission from depression with venlafaxine treatment in older adults. Am J Psychiatry. 2017;174:468–475. 26. Ahmed AT, Biernacka JM, Jenkins G, et al. Pharmacokinetic-pharmacodynamic interaction associated with venlafaxine-XR remission in patients with major depressive disorder with history of citalopram/escitalopram treatment failure. J Affect Disord. 2019;246:62–68. 27. Sánchez-Iglesias S, García-Solaesa V, García-Berrocal B, et al. Role of pharmacogenetics in improving the safety of psychiatric care by predicting the potential risks of mania in CYP2D6 poor metabolizers diagnosed with bipolar disorder. Medicine (Baltimore). 2016;95:e2473. 28. Malhi GS, Bell E, Morris G, et al. The Delay in Response to Antidepressant Therapy: A Window of Opportunity? Vol 54: SAGE Publications Sage UK: London, England; 2020:127–129. 29. Andreazza AC, Duong A, Young LT. Bipolar disorder as a mitochondrial disease. Biol Psychiatry. 2018;83:720–721. 30. Andreazza AC, Shao L, Wang J-F, et al. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch Gen Psychiatry. 2010;67:360–368. 31. Kato T. Neurobiological basis of bipolar disorder: mitochondrial dysfunction hypothesis and beyond. Schizophr Res. 2017;187:62–66. 32. Giménez-Palomo A, Dodd S, Anmella G, et al. The role of mitochondria in mood disorders: from physiology to pathophysiology and to treatment. Front Psych. 2021;12:546801. 33. Scaini G, Rezin GT, Carvalho AF, et al. Mitochondrial dysfunction in bipolar disorder: evidence, pathophysiology and translational implications. Neurosci Biobehav Rev. 2016;68:694–713. 34. Davey J, Turner RM, Clarke MJ, et al. Characteristics of meta-analyses and their component studies in the Cochrane database of systematic reviews: a cross-sectional, descriptive analysis. BMC Med Res Methodol. 2011;11:160. 35. von Hippel PT. The heterogeneity statistic I(2) can be biased in small meta-analyses. BMC Med Res Methodol. 2015;15:35–38. 36. Viktorin A, Lichtenstein P, Thase ME, et al. The risk of switch to mania in patients with bipolar disorder during treatment with an antidepressant alone and in combination with a mood stabilizer. Am J Psychiatry. 2014;171:1067–1073. 37. Gardea-Resendez M, Coombes BJ, Veldic M, et al. Antidepressants that increase mitochondrial energetics may elevate risk of treatment-emergent mania. Mol Psychiatry. 2022;28:1020–1026. 38. Malhi GS, Mann JJ. Depression. Lancet. 2018;392:2299–2312. 39. Mundo E, Cattaneo E, Russo M, et al. Clinical variables related to antidepressant-induced mania in bipolar disorder. J Affect Disord. 2006;92(2–3):227–230. 40. Perlis RH. A clinical risk stratification tool for predicting treatment resistance in major depressive disorder. Biol Psychiatry. 2013;74:7–14. 41. Nunes A, Ardau R, Berghöfer A, et al. Prediction of lithium response using clinical data. Acta Psychiatr Scand. 2020;141:131–141. 42. Maciukiewicz M, Marshe VS, Hauschild A-C, et al. GWAS-based machine learning approach to predict duloxetine response in major depressive disorder. J Psychiatr Res. 2018;99:62–68. 43. Bitencourt AGV, Gibbs P, Rossi Saccarelli C, et al. MRI-based machine learning radiomics can predict HER2 expression level and pathologic response after neoadjuvant therapy in HER2 overexpressing breast cancer. EBioMedicine. 2020;61:103042. 44. Farahmand S, Fernandez AI, Ahmed FS, et al. Deep learning trained on hematoxylin and eosin tumor region of interest predicts HER2 status and trastuzumab treatment response in HER2+ breast cancer. Mod Pathol. 2022;35:44–51. 45. Sammut S-J, Crispin-Ortuzar M, Chin S-F, et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature. 2022;601:623–629. 46. Kohavi R. A study of cross-validation and bootstrap for accuracy estimation and model selection. IJCAI. 1995;14:1137–1145. 47. Kim JH. Estimating classification error rate: repeated cross-validation, repeated hold-out and bootstrap. Comput Stat Data Anal. 2009;53:3735–3745.
留言 (0)