
記住我
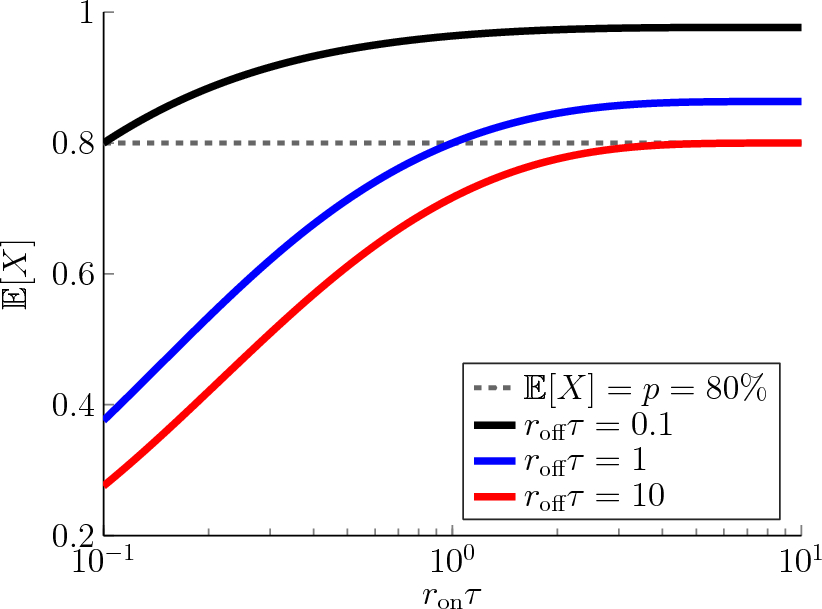
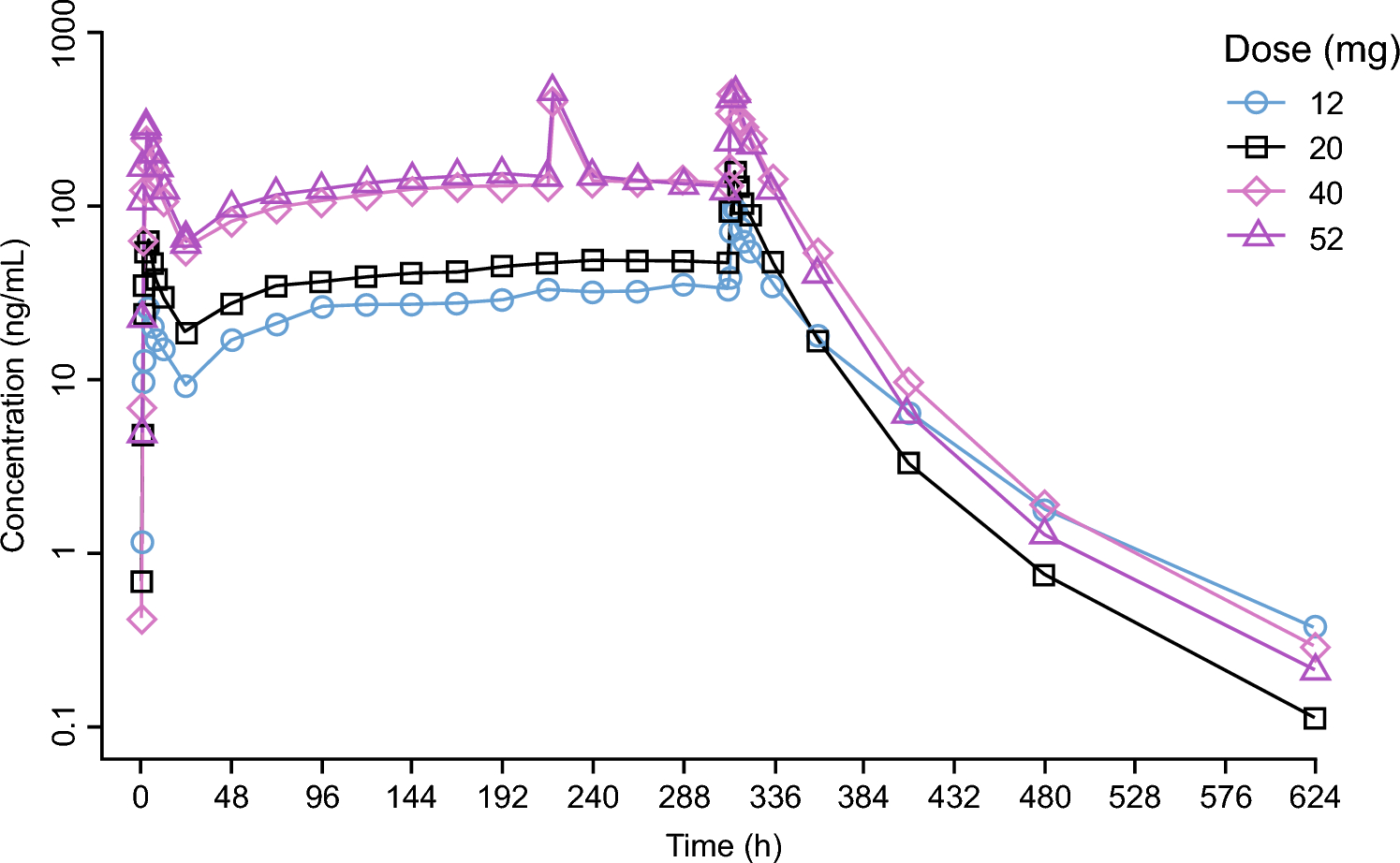
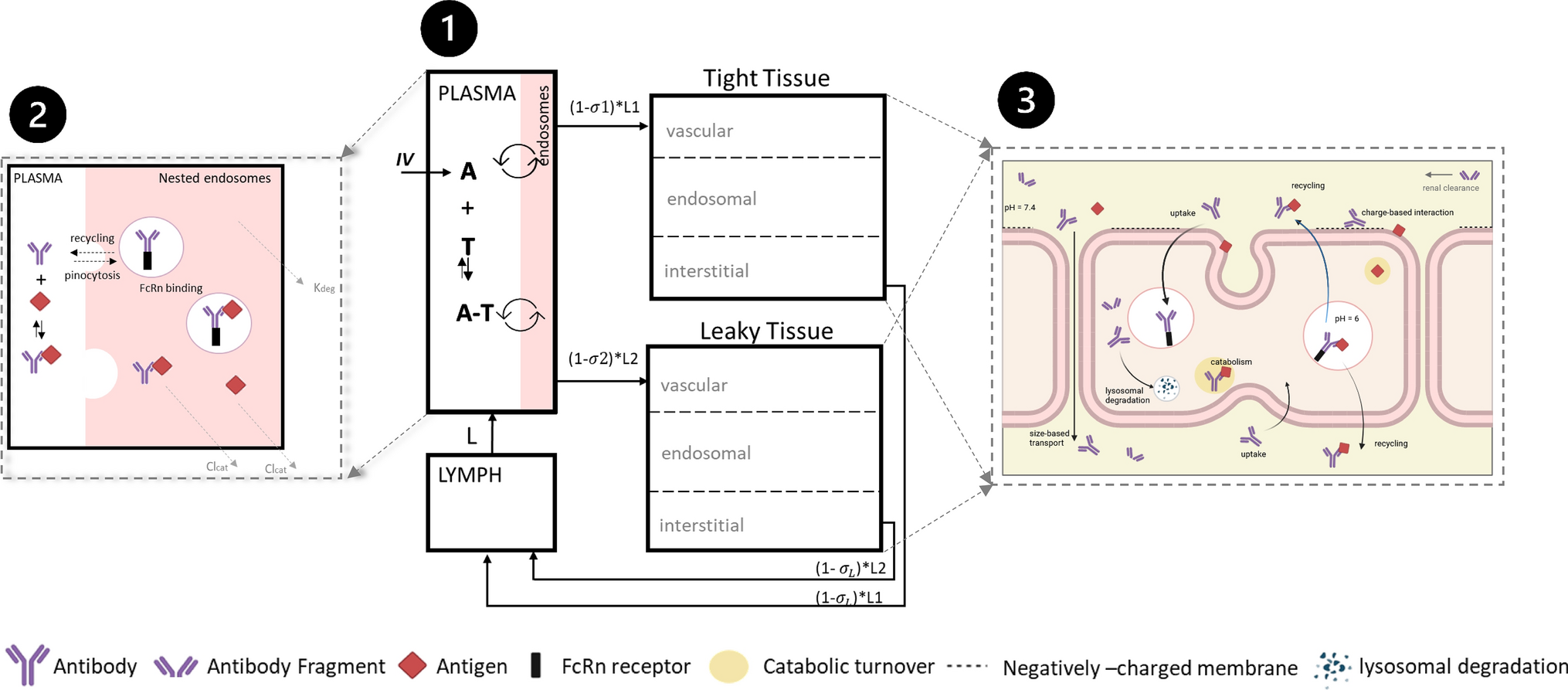


A PBPK model was developed for enfortumab vedotin using clinical data from phase 1 and 2 studies. The phase 1 dose-escalation/dose-expansion study of enfortumab vedotin (EV-101; NCT02091999) [16] was conducted in previously treated patients with Nectin-4–expressing metastatic UC and other malignant solid tumors. Single IV dose data of enfortumab vedotin 1.25 mg/kg were extracted from the study for model building and single IV dose data of enfortumab vedotin 1.0 mg/kg were used for single-dose model verification [16]. In the phase 2 single-cohort study of enfortumab vedotin (EV-201; NCT03219333), 125 patients with locally advanced/metastatic UC who were previously treated with platinum and PD-1/L1 inhibitor therapy received enfortumab vedotin 1.25 mg/kg. Multiple-dose data of enfortumab vedotin 1.25 mg/kg obtained from the study were used for multiple-dose verification [10]. Data from this study were split into a model development and model verification group.
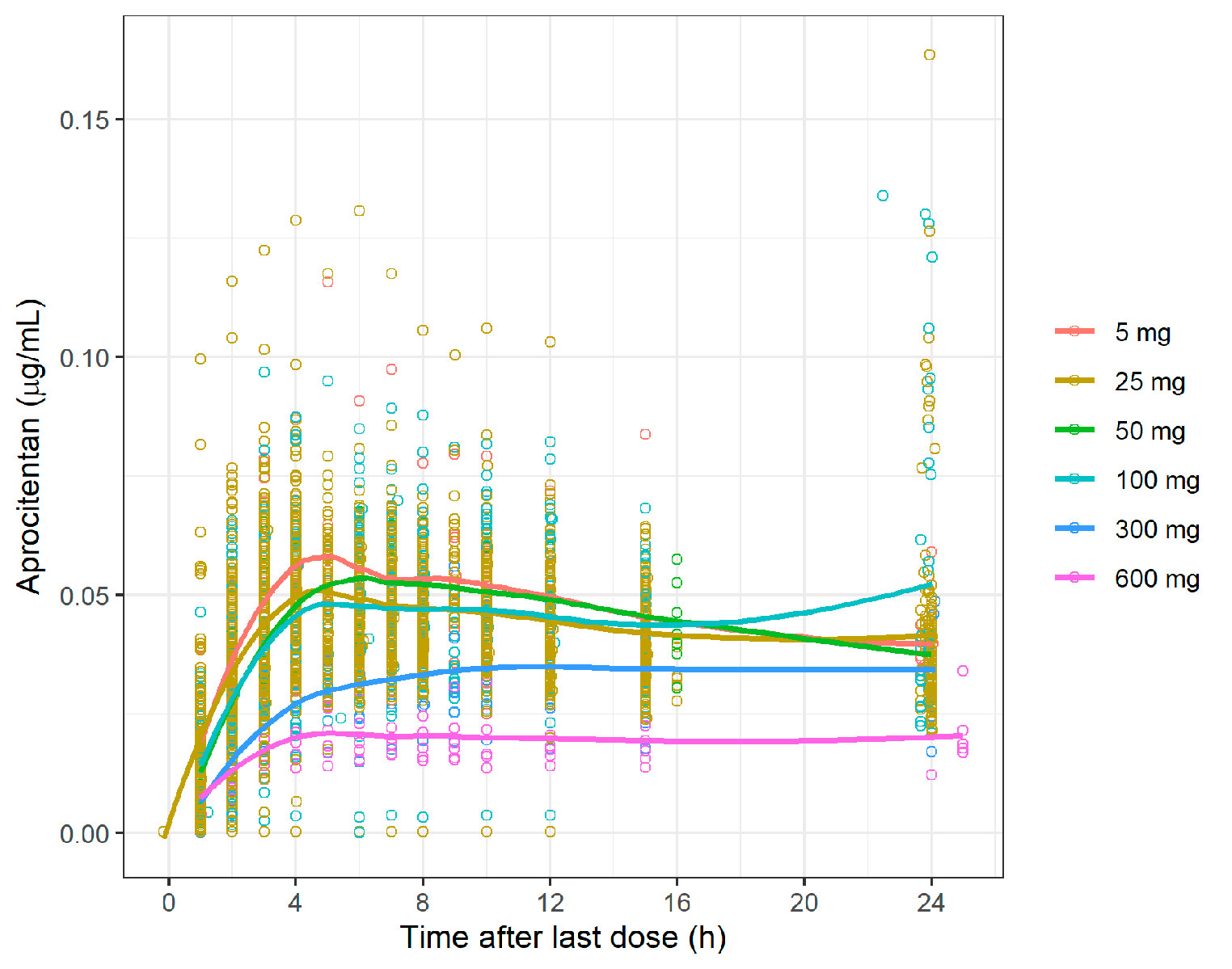
Brentuximab vedotin was used in the present analysis as a baseline comparison to enfortumab vedotin because of the availability of data, including a clinical DDI trial, and similarity in the average drug–antibody ratios between these compounds. Brentuximab vedotin data, which were available from the sponsor’s clinical pharmacology submission to the US Food and Drug Administration, were digitized for model development [11]. The PK data from a single IV dose of brentuximab vedotin 1.8 mg/kg administered over 30 min were used for model building. Data from a single IV dose of brentuximab vedotin 2.7 mg/kg were used for model verification. The clinical DDI study (NCT01026415) with brentuximab vedotin and a combined P-gp and CYP3A inhibitor or inducer was used for DDI verification [17]. For verification of P-gp–mediated interactions by ketoconazole and rifampin, clinical DDI data were obtained from prior publications [12, 13]. Data were extracted from the literature using GetData Graph Digitizer version 2.26. The process for PBPK model construction, verification, and application of enfortumab vedotin is depicted in Fig. 1.
Fig. 1
Process for PBPK model construction, verification, and application of enfortumab vedotin. DDI, drug–drug interaction; IV, intravenous; MMAE, monomethyl auristatin E; PBPK, physiologically based pharmacokinetics
Virtual trial designs for the enfortumab vedotin and brentuximab vedotin PBPK models closely resembled the actual clinical studies [16, 17, 26]; however, for enfortumab vedotin, the default numbers of patients in the Simcyp (Certara, Sheffield, UK) simulator (10 trials with 10 patients in each trial) were used. Specifics of the virtual trial design for model construction and verification for enfortumab vedotin and brentuximab vedotin can be found in the Supplementary Methods.
Model constructionThe ADC module provided within the Simcyp simulator version 19 was used for PBPK modeling (Fig. 2). For enfortumab vedotin, the PBPK model was built using the minimal PBPK modeling approach and, for MMAE, the full PBPK approach for small molecules was used (Tables 1 and 2). The neonatal Fc receptor (FcRn) dissociation constant was optimized for enfortumab vedotin based on clinical data because an in vitro experimental value was unavailable. It was determined that the FcRn dissociation constant would be different between brentuximab vedotin and enfortumab vedotin because of the difference in mAb and that the half-life of enfortumab vedotin (~ 3.6 days for the conjugated antibody) was shorter than brentuximab vedotin (~ 4 to 6 days). A lower binding affinity (higher dissociation) to FcRn was required for enfortumab vedotin to capture the ADC elimination time curve and production of MMAE. The source of unconjugated MMAE in the model was through catabolism, additional nonspecific plasma clearance, and deconjugation. Uptake clearance and recycling rate values were unknown for enfortumab vedotin or brentuximab vedotin; due to difficulties in measuring these parameters, default values provided within the Simcyp software were used.
Fig. 2
Schematic of (a) Simcyp simulator ADC module (b) linked to full PBPK model for small molecules. Reprinted with permission from Certara UK Limited. ADC, antibody-drug conjugate; IV, intravenous; PBPK, physiologically based pharmacokinetic model
Table 1 Input parameters of the PBPK model for enfortumab vedotin using the minimal PBPK model for ADC Table 2 Input parameters of the PBPK model for monomethyl auristatin E using the full PBPK model for small moleculesThe MMAE compound model was constructed using values obtained from a previous study of PBPK model building of brentuximab vedotin with some modifications [20], because the previous model was built using an older version of Simcyp that did not contain the ADC module. Use of the volume of distribution at steady state and CYP3A4 intrinsic clearance reported in the reference study led to an underestimation of MMAE exposure. Thus, these parameters were optimized for the ADC module and the enfortumab vedotin PBPK model. For the brentuximab vedotin PBPK model, ADC plasma clearance was optimized using clinically observed data from phase 1 studies with brentuximab vedotin administered at 1.8 mg/kg. The same MMAE compound file was utilized for enfortumab vedotin and brentuximab vedotin (Table S1).
Model verificationFor enfortumab vedotin, the PBPK model was verified by analyzing the ADC and MMAE concentration time profiles with single-dose (from the first dose within a cycle) enfortumab vedotin 1.0 mg/kg and multiple doses of enfortumab vedotin 1.25 mg/kg from phase 1 and 2 trials, respectively [10, 16]. For brentuximab vedotin, the PBPK model was verified by the ADC and MMAE concentration time profiles with the dose of brentuximab vedotin 2.7 mg/kg from the clinical pharmacology data submitted to the US Food and Drug Administration [26].
Model verification of drug interactions with brentuximab vedotin was conducted with a combined P-gp and CYP3A4 inhibitor or inducer and CYP3A4 substrate. The simulated plasma concentration profiles, ratio of AUC from time 0 extrapolated to infinity (AUCinf), and ratio of maximum serum concentration (Cmax) of ADC and MMAE from brentuximab vedotin 1.2 mg/kg with ketoconazole 400 mg oral daily or brentuximab vedotin 1.8 mg/kg with rifampin 600 mg oral daily or a single IV dose of midazolam 1 mg were compared with clinical PK data from a previously published study [17]. Rifampin simulations were performed with the fold-increase of the P-gp transporter relative activity factor value applied to MMAE, as previously described.
For the cancer population, the distributions of simulated values for age, body weight, plasma albumin value, and hematocrit level were compared with those observed from all patients in the phase 1 study [16] for verification of the Simcyp simulator–provided cancer population modification.
Model for drug–drug interaction simulationsThe verified PBPK model was used to evaluate the interaction of enfortumab vedotin with ketoconazole, rifampin, midazolam, and digoxin. For the DDI simulation studies, the clinical brentuximab vedotin DDI data were leveraged to verify the applicability of the brentuximab vedotin PBPK model in predicting DDIs observed clinically. This information was then used for enfortumab vedotin to predict the exposure of MMAE in several DDI simulations.
To analyze effects of the combined P-gp and CYP3A4 inhibitor and inducer on enfortumab vedotin and brentuximab vedotin, the Simcyp simulator–provided PBPK models for ketoconazole and rifampin were slightly modified. An inhibitory constant (Ki) for the P-gp transport for intestine and liver compartments was analyzed and incorporated for both models; for ketoconazole, a P-gp inhibition KI value of 0.67 µM was added to the ketoconazole PBPK compound model file; for rifampin, a KI value of 4.3 µM was added. No other parameters were altered.
To analyze the inhibitory effects of enfortumab vedotin and brentuximab vedotin on CYP3A4 and P-gp substrates, the Simcyp simulator PBPK models for midazolam and digoxin were used for enfortumab vedotin and brentuximab vedotin DDI simulations. The model for midazolam was used without modification. For simulations with rifampin, the relative activity factor for P-gp transport within the digoxin compound file was increased to represent transporter induction. Rifampin simulations were performed twice: once with the increased relative activity value contained within the digoxin compound file, then again with the baseline relative activity value and rifampin dose set to 0. Of note, the state of the simulator at the time of analysis did not support transporter induction for DDI simulation; therefore, the effect of P-gp induction by rifampin was manually implemented.
The difference between enfortumab vedotin and brentuximab vedotin populations was hypothetically linked to the included cancer population (solid tumor vs. blood cancer) within the clinical studies [16, 26]. The Simcyp cancer population model was used without modification for all brentuximab vedotin simulations. For simulations of enfortumab vedotin, the population was slightly modified and the influence of the tissue–volume scaling factor for plasma on the ADC plasma concentrations was investigated. For digoxin simulations, the Simcyp simulator–containing PBPK model for the healthy volunteer population was used without modifications.
Application of the drug–drug interaction simulation and sensitivity analysisThe verified enfortumab vedotin PBPK model was employed to simulate interactions between enfortumab vedotin and rifampin, ketoconazole, midazolam, and digoxin using the virtual trial design (Table S2). Sensitivity analysis was conducted to evaluate the uncertainty of P-gp (biliary) versus CYP3A4 contribution on elimination of MMAE. The drug interactions between enfortumab vedotin and ketoconazole or rifampin were used to analyze the effect of the elimination pathway on the GMRs of MMAE Cmax and AUC from time 0 to last quantifiable concentration (AUClast; information regarding times for AUClast appears in Table S2). Simulations were run for a sufficient time duration for the AUClast ratio to be equivalent to the AUCinf ratio.
Statistical analysisSummary statistics of simulated Cmax, AUClast, AUCinf, AUC from time 0 to day 7 (AUCd0–7), AUC from time 0 to day 14 (AUCd0–14), Cmax ratio, and AUCinf/last ratio were calculated by noncompartmental analysis. For DDI simulations involving rifampin, midazolam, and digoxin, geometric mean ratios (GMRs) were calculated for Cmax and AUClast (reported instead of AUCinf due to limitations in calculating AUCinf for some simulations). The GMR was calculated from the 2 simulations using R-Studio (R-Studio, Boston, MA).
留言 (0)