
記住我



Dermatofibrosarcoma protuberans (DFSP) is a skin tumor that involves cutaneous soft tissues. This relatively rare tumor typically presents as a slowly progressive plaque on the trunk of young adults. DFSP has an expansive differential diagnosis (Table 1). It typically resembles a scar that slowly grows for months to years. This low-grade tumor has a high likelihood of local recurrence unless it is completely excised. Because of its rarity and subtle progression, this skin cancer will often go undiagnosed for years (Tanwar et al., 2021). Over time, DFSP can become invasive into the subcutaneous fat, muscle, and fascia. Studies have found a chromosomal translocation that is associated with over 90% of DFSP cases and is thought to be key to the development of this tumor. This translocation promotes growth of the tumor through platelet-derived growth factor (PDGF) overproduction (Simon et al., 1997; Sirvent et al., 2003).
TABLE 1 - Differential Diagnosis of Dermatofibrosarcoma Protuberans Differential Diagnosis - Basal cell carcinomaThe incidence of DFSP in the United States has been estimated to be 0.8–4.5 per million persons per year (Gloster, 1996; Kreicher et al., 2016; Rouhani et al., 2008). In a 2016 report on the incidence and survival of DFSP using all 18 registries from the Surveillance, Epidemiology, and End Results database through 2010, Kreicher et al. (2016) found that the overall incidence of DFSP was 4.1 per million person-years and was steady over the decade. They also found the incidence to be 1.14 times higher among women compared with men. The incidence among Blacks was nearly 2 times the incidence among whites. The trunk was the most common location, accounting for 41.6% of cases. The incidence of DFSP is highest in the 20- to 39-year age bracket (42% of cases) and the 40- to 59-year age bracket (37.7% of cases). Five-year and 10-year relative survival rates were found to be at 99.8% and 99.1%, respectively. Similar findings were found by Rouhani et al. in a 2008 report on population-based cancer incidence data from 1992 through 2004 among residents of the 13 Surveillance, Epidemiology, and End Results Program registries (Rouhani et al., 2008).
PathogenesisOver 90% of DFSP cases are characterized by a translocation t(17;22). This translocation results in the fusion of the PDGF beta polypeptide (PDGFB gene) with the collagen type 1A1 (COL1A1) gene (Maki et al., 2002; Sandberg & Bridge, 2003). This fusion leads to overproduction of PDGF and continuous activation of the PDGFB receptor. This ultimately leads to increased cell proliferation and tumor formation. Although 34 different variants of this fusion gene have been recognized, there appears to be no association between the specific variant and the clinical characteristics of DFSP (Llombart et al., 2009). Two notable subtypes of DFSP that are well studied are the Bednar pigmented variant (<5% of cases) and the higher-grade fibrosarcomatous variant (5%–15% of cases). There are a few reasons why this fusion gene is clinically relevant. First, this fusion is found in the vast majority of DFSP cases and likely is fundamental to tumor development. Next, fluorescence in situ hybridization (FISH) and reverse transcription polymerase chain reaction (RT-PCR) can be used to detect these PDGF-B/COL1A1 fusion transcripts. Detecting these fusion transcripts can be used both for diagnostic purposes and for choosing the most appropriate treatment option (Patel et al., 2008). Last, the constitutive activation of PDGFB receptors that results from this fusion protein provides a potential target for future treatment modalities.
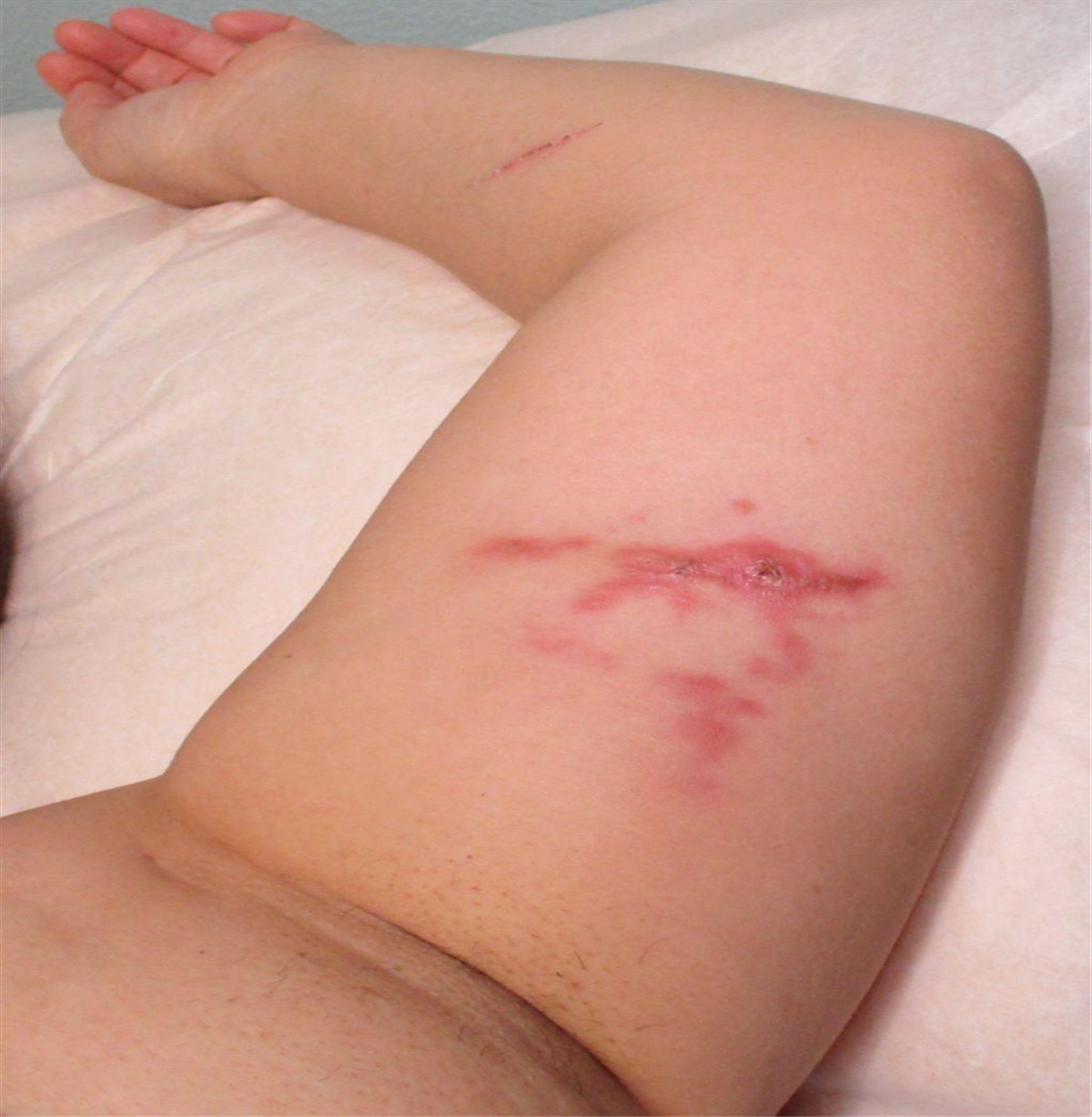
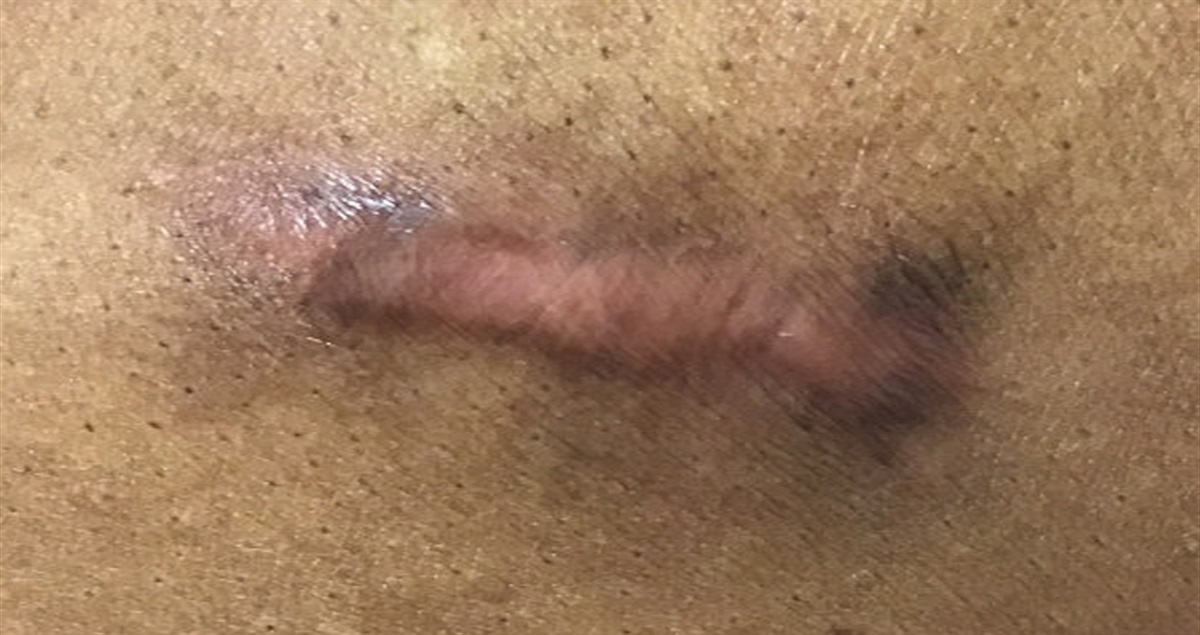
Clinical FeaturesDFSP is often asymptomatic initially. In most cases, the tumor has been present for many years by the time it is diagnosed (Mentzel et al., 1998). This tumor classically arises from the dermis as a plaque that grows progressively over the course of months to years. It can rarely present as a cutaneous nodule that is firm. The tumor arises as a pink, red, or violet plaque. These plaques can become telangiectatic over time with small, widened blood vessels becoming visible on the skin's surface (Lindner et al., 1999). The most common sites where lesions arise include the trunk (41.6%), upper limb (21.2%), lower limb (20.8%), and head (12.9%). Rarely, DFSP can arise from a tattoo or preexisting scar (De Antoni et al., 2020; Hirano & Torosky, 2012). Incidence rates appear to be highest in patients who are in their 30s (Bowne et al., 2000; Kreicher et al., 2016; Lindner et al., 1999). The tumor is commonly fixed to the dermis but may involve deeper subcutaneous tissues later in the disease course. Over time, the tumor may become a raised, firm nodule (Figure 1). Once the tumor takes a nodular form, the overlying skin may become shiny or ulcerated and may be painful.
 FIGURE 1.:
FIGURE 1.: Dermatofibrosarcoma protuberans presenting as a firm, pink nodule on the upper back of an 8-year-old child.
At the time of diagnosis, DFSP remains superficial and has a diameter of less than 5 cm in most cases (Bowne et al., 2000). On occasion, these tumors can grow to become very large. If the tumor remains untreated, its growth generally accelerates. Accelerated growth can also occur during pregnancy (Har-Shai et al., 1993; Parlette et al., 1999).
The metastatic rate for low-grade DFSP is thought to be about 1% (Cai et al., 2012; Lemm et al., 2009). Malignancy is more common in patients who have had tumor recurrence and patients with the more malignant fibrosarcomatous variant of DFSP. Distant metastasis appears to primarily occur via the bloodstream, affecting the lungs most commonly. Spread to regional lymph nodes appears to be relatively rare, with regional lymph node metastases being observed in only three of the 260 patients with DFSP involved in a 2012 study (Cai et al., 2012).
DIAGNOSIS DermoscopyDermoscopy can be useful in the diagnostic workup of lesions if DFSP is suspected. DFSP should be suspected in all patients who present with a firm, slow-growing nodule. A 2013 study by Bernard et al. sought to describe the main dermoscopy findings in DFSP (Bernard et al., 2013). They did this by determining the frequency of six predefined dermoscopy findings among a group of 15 cases of biopsy-proven DFSP. The most common observed finding was a pigmented network, seen in 87% of lesions. The next most frequently observed findings were vessel structures (seen in 80% of lesions), structureless light-brown areas (73% of lesions), shiny white streaks (67% of lesions), pink background coloration (67% of lesions), and structureless hypopigmented/depigmented areas (60% of lesions). The median number of the predefined dermoscopy findings found per lesion was notably high at 4. Although none of the predefined dermoscopy findings are specific to DFSP, a collection of several of these findings should raise suspicion about this tumor. Importantly, no dermoscopy differences were found between low-grade DFSP and the fibrosarcomatous variant of DFSP. Furthermore, no differences were found on dermoscopy between primary lesions and lesions that were recurrent.
Dermoscopy can be utilized in differentiating DFSP from other similar lesions. Dermatofibromas are pedunculated lesions more typically seen in extremities. Cutaneous neurofibromas are skin-colored, rubbery nodules that grow slowly. Solitary fibrous tumors are slow-growing, skin-colored masses. Although dermoscopy can help distinguish lesions that warrant biopsy from those that are more likely benign, new and evolving skin lesions warrant biopsy in most cases.
HistopathologyAlthough dermoscopy can be helpful when DFSP is suspected, the diagnosis of this skin cancer requires an incisional or core needle biopsy. Of note, fine needle aspiration is not used for diagnosing primary DFSP but can be used for diagnosing recurrent disease.
DFSP appears poorly circumscribed and often infiltrates the full thickness of the dermis, destroying preexisting structures (Llombart et al., 2013). The tumor is composed of a dense array of monomorphic cells with spindle-shaped nuclei embedded in collagen. The tumor cells have large nuclei, rare mitotic figures, and limited size and shape variability. These proliferating cells are often arranged in fascicles that are irregular and interwoven. Higher cellularity is often seen centrally in the tumor, with peripheral areas being less cellular. This results in a storiform or irregularly whirled pattern (Llombart et al., 2013). The tumor cells can be arranged around a central hub, like the spokes of a wheel. Small amounts of mucin may be present beneath the epidermis. Often, the epidermis that overlies the tumor is thin with flattened rete ridges. Moderate epidermal thickening can less commonly be present.
The main characteristic of DFSP is its capacity to invade tissues surrounding the tumor. Invasion of surrounding tissue occurs through tentacle-like projections. Invasion of surrounding fat is said to occur in a “honeycomb” pattern of fat surrounded by infiltrating tumor cells or in a “sandwich” pattern or spindle cell layers that run parallel to the surface of the skin (Bague & Folpe, 2008; Zelger et al., 1994). The histologic appearance of these invading projections often resembles normal fibrous tracts, possibly playing a role in the recurrence of DFSP in patients who were said to have excision with wide margins.
The histological appearance of the fibrosarcomatous variant of DFSP has a characteristic fascicular or herringbone architecture. This variant is generally more hypercellular, pleomorphic, mitotically active, and aggressive. It is also associated with an expansive pattern (Connelly & Evans, 1992; Llombart et al., 2013; Sanz-Trelles et al., 1998).
The histology of the Bednar pigmented variant of DFSP is characterized by a storiform pattern of spindle cells with melanin-containing dendritic cells (Llombart et al., 2013). The histologic population of melanin-containing cells is variable among Bednar tumors. The density of melanin-containing cells correlates with the appearance of the tumor grossly, with some tumors having such a low density of these cells that they do not grossly appear pigmented and can only be defined as Bednar tumors following biopsy.
ImmunohistochemistryDFSP characteristically stains positive for CD34 and negative for Factor XIIIa (Caccavale et al., 2022). Staining positive for CD34 helps differentiate DFSP from other soft tissue tumors, such as dermatofibroma. Approximately 80%–100% of DFSP tumors stain positive for CD34, whereas about 95% of dermatofibromas stain negatively for CD34 (Abenoza & Lillemoe, 1993; Haycox et al., 1997; Kamino et al., 1992; Zelger et al., 1994). Importantly, the sarcomatous component of the fibrosarcomatous variant of DFSP does not stain positive for CD34, which can be helpful in differentiating this variant from other forms of DFSP.
Molecular TestingFISH and RT-PCR techniques can both be useful in diagnosing DFSP by detecting the translocation t(17;22). Although the use of these molecular techniques is often not required for the diagnosis of DFSP, detecting this translocation is useful for predicting how a patient will respond to treatment. Patients with this translocation respond particularly well to imatinib. This translocation, resulting in overproduction of PDGF and continuous activation of the PDGFB receptor, is present in over 90% of DFSP cases. Both techniques have been shown to have a specificity of 100% for detecting the PDGFB/COL1A1 transcript. FISH, however, has been shown to be more sensitive (90% sensitive) compared with RT-PCR (72% sensitive) for detecting this transcript (Salgado et al., 2011).
StagingAlthough there are several staging systems used worldwide, many clinicians in the United States stage DFSP using the American Musculoskeletal Tumor Society staging system. This staging system was first defined by Enneking et al. in 1980 and is based on histological grade (low vs. high) and anatomic compartment confinement (intercompartmental vs. extracompartmental). Stage I is defined in this grading system as low grade, with Stage IA representing intracompartmental confinement and Stage IB representing extracompartmental tumors. Stage II is defined by being high grade, with Stage IIA representing intracompartmental confinement and Stage IIB representing extracompartmental tumors. Stage III is reserved for tumors with systemic or regional metastases (Enneking et al., 1980).
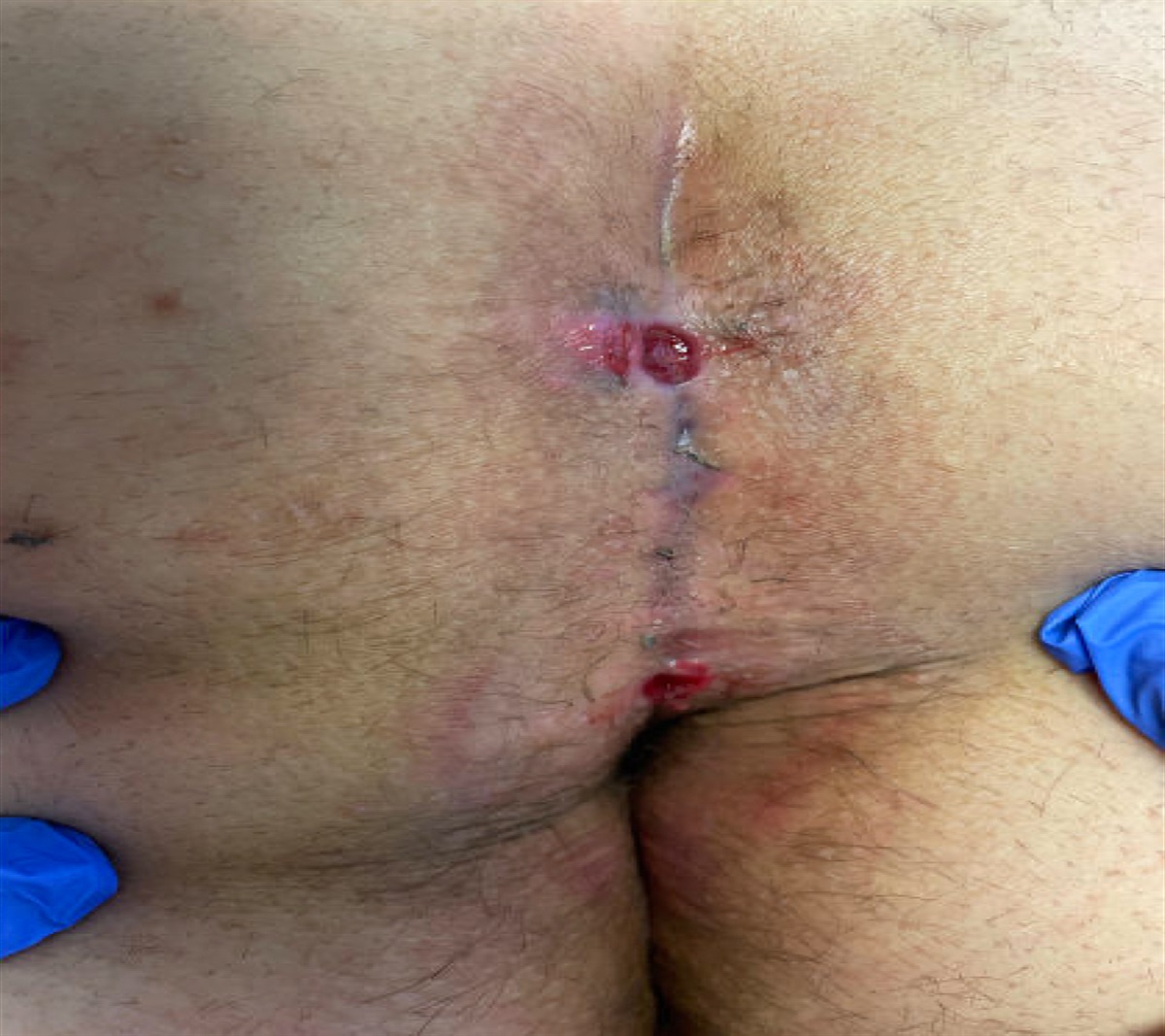
TREATMENTThe preferred treatment for DFSP is resection with negative margins. The size and location of the tumor are both important factors when determining the most appropriate surgical procedure. It is very important to achieve negative margins because failure to do so increases the risk of locally recurrent disease that is associated with a higher risk of more aggressive and metastatic disease. Magnetic resonance imaging can be helpful preoperatively to better define tumor extension before surgery. The preferred treatment option is Mohs micrographic surgery (Paradisi et al., 2008; Shah et al., 2021). This technique is particularly useful in DFSP because clear histopathologic margins are confirmed during the surgical procedure. Recurrence rates after Mohs micrographic surgery are around 1%–1.5% (Foroozan et al., 2012; Martin et al., 2022). If Mohs surgery is not available, wide local excision may be performed with 2- to 4-cm margins. Even with wide local excision and clear margins, the local recurrence of DFSP is 4%–9% (Foroozan et al., 2012; Martin et al., 2022). Careful clinical examination of regional lymph nodes in the preoperative and postoperative periods can be useful for conservatively assessing lymph node metastasis in patients with diagnosed DFSP (Mavili et al., 1994). Lymph node dissection is not generally recommended.
Imatinib mesylate is a medication that is FDA approved for the treatment of DFSP. Imatinib has been shown to be safe and effective for use in adult patients who have unresectable, recurrent, and/or metastatic DFSP. This medication is a tyrosine kinase inhibitor that competitively inhibits adenosine triphosphate (ATP) binding to the PDGFB receptor. This promotes apoptosis and limits tumor growth. Patients should be screened for the translocation t(17;22) before treatment, as patients with this translocation respond particularly well to imatinib mesylate (65% response rate).
Radiation is rarely used as the primary treatment for DFSP but can be used as adjuvant therapy in some cases. Radiation has been shown to decrease the risk of recurrence when used in addition to surgery (Dagan et al., 2005; Haas et al., 1997; Sun et al., 2000). For patients with lymph node involvement, lymphadenectomy is recommended. Surgical resection may be appropriate for some patients with isolated sites of metastasis.
FOLLOW-UPBecause of the recurrent nature of DFSP, close clinical follow-up after treatment is important. Although recurrence rates are highest within 3 years of treatment, 25%–30% of recurrent DFSP cases occur more than 5 years after treatment. Because of this significant risk for recurrence, it is recommended that patients be seen every 6 months for the first 3 years and then annually for the rest of their life (Llombart et al., 2013). Routine imaging is generally not recommended for patients with DFSP unless symptoms suggest metastasis. Some symptoms that may suggest DFSP metastasis include fatigue, weight loss, shortness of breath, and loss of appetite. Some sources suggest routine imaging in patients with the fibrosarcomatous variant of DFSP because of its higher potential for metastasis.
KEY CONCEPTS DFSP is a relatively rare, soft tissue tumor that arises from the dermis. Although DFSP is generally low-to-intermediate grade, it has a high propensity for local recurrence unless completely excised. Over 90% of DFSP cases are characterized by a translocation t(17;22) that results in overproduction of PDGF and continuous activation of the PDGFB receptor. This ultimately leads to increased cell proliferation and tumor formation. This tumor classically arises from the dermis as a plaque that grows progressively over the course of months to years. This tumor arises as a pink or violet–red plaque that can become telangiectatic over time. This tumor most commonly arises on the patient's trunk and is most common in patients in their 30s. The definitive diagnosis of DFSP requires an incisional or core needle biopsy. The preferred initial treatment option for localized DFSP is resection with negative margins. Mohs micrographic surgery is associated with recurrence rates of 1%–1.5%. Imatinib mesylate is an FDA-approved medication that is safe and effective for adult patients with unresectable, recurrent, and/or metastatic DFSP. Because of the significant risk for tumor recurrence, it is recommended that patients be seen every 6 months for the first 3 years after treatment and then annually for the rest of their life. Five-year and 10-year relative survival rates of DFSP have been estimated to be 99.8% and 99.1%, respectively.
 REFERENCES
Abenoza P., Lillemoe T. (1993). CD34 and factor XIIIa in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans. The American Journal of Dermatopathology, 15(5), 429–434. 10.1097/00000372-199310000-00003
Bague S., Folpe A. L. (2008). Dermatofibrosarcoma protuberans presenting as a subcutaneous mass: A clinicopathological study of 15 cases with exclusive or near-exclusive subcutaneous involvement. The American Journal of Dermatopathology, 30(4), 327–332. 10.1097/DAD.0b013e31817d32b2
Bernard J., Poulalhon N., Argenziano G., Debarbieux S., Dalle S., Thomas L. (2013). Dermoscopy of dermatofibrosarcoma protuberans: A study of 15 cases. British Journal of Dermatology, 169(1), 85–90. 10.1111/bjd.12318
Bowne W. B., Antonescu C. R., Leung D. H., Katz S. C., Hawkins W. G., Woodruff J. M., Brennan M. F., Lewis J. J. (2000). Dermatofibrosarcoma protuberans: A clinicopathologic analysis of patients treated and followed at a single institution. Cancer, 88(12), 2711–2720.
Caccavale S., Martins Basso A., Vitiello P., Ronchi A., Sica A., Verolino P., Toncic R. J., Argenziano G. (2022). Dermatofibrosarcoma protuberans: Experience at a third-level referral center. Dermatology Practical & Conceptual, 12(1), e2022033. 10.5826/dpc.1201a33
Cai H., Wang Y., Wu J., Shi Y. (2012). Dermatofibrosarcoma protuberans: Clinical diagnoses and treatment results of 260 cases in China. Journal of Surgical Oncology, 105(2), 142–148. 10.1002/jso.22000
Connelly J. H., Evans H. L. (1992). Dermatofibrosarcoma protuberans. A clinicopathologic review with emphasis on fibrosarcomatous areas. American Journal of Surgical Pathology, 16(10), 921–925.
Dagan R., Morris C. G., Zlotecki R. A., Scarborough M. T., Mendenhall W. M. (2005). Radiotherapy in the treatment of dermatofibrosarcoma protuberans. American Journal of Clinical Oncology, 28(6), 537–539. 10.1097/01.coc.0000171278.69291.64
De Antoni E., Brambullo T., Pescarini E., Salmaso R., Bassetto F., Vindigni V. (2020). Dermatofibrosarcoma protuberans on tattooed skin: A case report. Advances in Skin & Wound Care, 33(2), 104–108. 10.1097/01.ASW.0000613548.11947.b4
Enneking W. F., Spanier S. S., Goodman M. A. (1980). A system for the surgical staging of musculoskeletal sarcoma. Clinical Orthopaedics and Related Research, (153), 106–120.
Foroozan M., Sei J. F., Amini M., Beauchet A., Saiag P. (2012). Efficacy of Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans: Systematic review. Archives of Dermatology, 148(9), 1055–1063. 10.1001/archdermatol.2012.1440
Gloster H. M. (1996). Dermatofibrosarcoma protuberans. Journal of the American Academy of Dermatology, 35(3, Pt 1), 355–374. 10.1016/s0190-9622(96)90597-6
Haas R. L., Keus R. B., Loftus B. M., Rutgers E. J., van Coevorden F., Bartelink H. (1997). The role of radiotherapy in the local management of dermatofibrosarcoma protuberans. Soft Tissue Tumours Working Group. European Journal of Cancer, 33(7), 1055–1060. 10.1016/s0959-8049(97)00026-9
Har-Shai Y., Govrin-Yehudain J., Ullmann Y., Kerner H., Cohen H. I., Lichtig C., Bergman R., Cohen A., Kuten A., Friedman-Birnbaum R. (1993). Dermatofibrosarcoma protuberans appearing during pregnancy. Annals of Plastic Surgery, 31(1), 91–93.
Haycox C. L., Odland P. B., Olbricht S. M., Piepkorn M. (1997). Immunohistochemical characterization of dermatofibrosarcoma protuberans with practical applications for diagnosis and treatment. Journal of the American Academy of Dermatology, 37(3, Pt 1), 438–444. 10.1016/s0190-9622(97)70146-4
Hirano S. A., Torosky C. M. (2012). Dermatofibrosarcoma protuberans arising at a Rho(D) immune globulin injection site. Cutis, 90(5), 233–234.
Kamino H., Reddy V. B., Gero M., Greco M. A. (1992). Dermatomyofibroma. A benign cutaneous, plaque-like proliferation of fibroblasts and myofibroblasts in young adults. Journal of Cutaneous Pathology, 19(2), 85–93. 10.1111/j.1600-0560.1992.tb01348.x
Kreicher K. L., Kurlander D. E., Gittleman H. R., Barnholtz-Sloan J. S., Bordeaux J. S. (2016). Incidence and survival of primary dermatofibrosarcoma protuberans in the United States. Dermatologic Surgery, 42(Suppl 1), S24–S31. 10.1097/DSS.0000000000000300
Lemm D., Mügge L. O., Mentzel T., Höffken K. (2009). Current treatment options in dermatofibrosarcoma protuberans. Journal of Cancer Research and Clinical Oncology, 135(5), 653–665. 10.1007/s00432-009-0550-3
Lindner N. J., Scarborough M. T., Powell G. J., Spanier S., Enneking W. F. (1999). Revision surgery in dermatofibrosarcoma protuberans of the trunk and extremities. European Journal of Surgical Oncology, 25(4), 392–397. 10.1053/ejso.1999.0663
Llombart B., Sanmartín O., López-Guerrero J. A., Monteagudo C., Serra C., Requena C., Poveda A., Vistós J. L., Almenar S., Llombart-Bosch A., Guillén C. (2009). Dermatofibrosarcoma protuberans: Clinical, pathological, and genetic (COL1A1-PDGFB) study with therapeutic implications. Histopathology, 54(7), 860–872. 10.1111/j.1365-2559.2009.03310.x
Llombart B., Serra-Guillén C., Monteagudo C., López Guerrero J. A., Sanmartín O. (2013). Dermatofibrosarcoma protuberans: A comprehensive review and update on diagnosis and management. Seminars in Diagnostic Pathology, 30(1), 13–28. 10.1053/j.semdp.2012.01.002
Maki R. G., Awan R. A., Dixon R. H., Jhanwar S., Antonescu C. R. (2002). Differential sensitivity to imatinib of 2 patients with metastatic sarcoma arising from dermatofibrosarcoma protuberans. International Journal of Cancer, 100(6), 623–626. 10.1002/ijc.10535
Martin E. C. S., Vyas K. S., Batbold S., Erwin P. J., Brewer J. D. (2022). Dermatofibrosarcoma protuberans recurrence after wide local excision versus Mohs micrographic surgery: A systematic review and meta-analysis. Dermatologic Surgery, 48(5), 479–485. 10.1097/DSS.0000000000003411
Mavili M. E., Gursu K. G., Gokoz A. (1994). Dermatofibrosarcoma with lymph node involvement. Annals of Plastic Surgery, 32(4), 438–440. 10.1097/00000637-199404000-00022
Mentzel T., Beham A., Katenkamp D., Dei Tos A. P., Fletcher C. D. (1998). Fibrosarcomatous ("high-grade") dermatofibrosarcoma protuberans: Clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. American Journal of Surgical Pathology, 22(5), 576–587. 10.1097/00000478-199805000-00009
Paradisi A., Abeni D., Rusciani A., Cigna E., Wolter M., Scuderi N., Kaufmann R., Podda M. (2008). Dermatofibrosarcoma protuberans: Wide local excision vs. Mohs micrographic surgery. Cancer Treatment Reviews, 34(8), 728–736. 10.1016/j.ctrv.2008.06.002
Parlette L. E., Smith C. K., Germain L. M., Rolfe C. A., Skelton H. (1999). Accelerated growth of dermatofibrosarcoma protuberans during pregnancy. Journal of the American Academy of Dermatology, 41(5, Pt 1), 778–783. 10.1016/s0190-9622(99)70023-x
Patel K. U., Szabo S. S., Hernandez V. S., Prieto V. G., Abruzzo L. V., Lazar A. J., López-Terrada D. (2008). Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Human Pathology, 39(2), 184–193. 10.1016/j.humpath.2007.06.009
Rouhani P., Fletcher C. D., Devesa S. S., Toro J. R. (2008). Cutaneous soft tissue sarcoma incidence patterns in the U.S.: An analysis of 12,114 cases. Cancer, 113(3), 616–627. 10.1002/cncr.23571
Salgado R., Llombart B., M Pujol R., Fernández-Serra A., Sanmartín O., Toll A., Rubio L., Segura S., Barranco C., Serra-Guillén C., Yébenes M., Salido M., Traves V., Monteagudo C., Sáez E., Hernández T., de Álava E., Llombart-Bosch A., Solé F., Guillén C., Espinet B., López-Guerrero J. A. (2011). Molecular diagnosis of dermatofibrosarcoma protuberans: A comparison between reverse transcriptase-polymerase chain reaction and fluorescence in situ hybridization methodologies. Genes Chromosomes & Cancer, 50(7), 510–517. 10.1002/gcc.20874
Sandberg A. A., Bridge J. A. (2003). Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors. Dermatofibrosarcoma protuberans and giant cell fibroblastoma. Cancer Genetics, 140(1), 1–12. 10.1016/s0165-4608(02)00848-8
Sanz-Trelles A., Ayala-Carbonero A., Rodrigo-Fernández I., Weil-Lara B. (1998). Leiomyomatous nodules and bundles of vascular origin in the fibrosarcomatous variant of dermatofibrosarcoma protuberans. Journal of Cutaneous Pathology, 25(1), 44–49. 10.1111/j.1600-0560.1998.tb01688.x
Shah A., Tassavor M., Sharma S., Torbeck R. (2021). The various treatment modalities of dermatofibrosarcoma protuberans. Dermatology Online Journal, 27(6). 10.5070/D327654070
Simon M. P., Pedeutour F., Sirvent N., Grosgeorge J., Minoletti F., Coindre J. M., Terrier-Lacombe M. J., Mandahl N., Craver R. D., Blin N., Sozzi G., Turc-Carel C., O'Brien K. P., Kedra D., Fransson I., Guilbaud C., Dumanski J. P. (1997). Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nature Genetics, 15(1), 95–98. 10.1038/ng0197-95
Sirvent N., Maire G., Pedeutour F. (2003). Genetics of dermatofibrosarcoma protuberans family of tumors: From ring chromosomes to tyrosine kinase inhibitor treatment. Genes Chromosomes & Cancer, 37(1), 1–19. 10.1002/gcc.10202
Sun L. M., Wang C. J., Huang C. C., Leung S. W., Chen H. C., Fang F. M., Huang E. Y., Lee S. P. (2000). Dermatofibrosarcoma protuberans: Treatment results of 35 cases. Radiotherapy and Oncology, 57(2), 175–181. 10.1016/s0167-8140(00)00228-0
Tanwar P., Singh A., Pratap S., Rattan A., Minhas S. S. (2021). Dermatofibrosarcoma—An uncommon entity, commonly mismanaged: A case report. Internation Journal of Surgical Case Reports, 87, 106385. 10.1016/j.ijscr.2021.106385
Zelger B., Sidoroff A., Stanzl U., Fritsch P. O., Ofner D., Jasani B., Schmid K. W. (1994). Deep penetrating dermatofibroma versus dermatofibrosarcoma protuberans. A clinicopathologic comparison. American Journal of Surgical Pathology, 18(7), 677–686. 10.1097/00000478-199407000-00003
REFERENCES
Abenoza P., Lillemoe T. (1993). CD34 and factor XIIIa in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans. The American Journal of Dermatopathology, 15(5), 429–434. 10.1097/00000372-199310000-00003
Bague S., Folpe A. L. (2008). Dermatofibrosarcoma protuberans presenting as a subcutaneous mass: A clinicopathological study of 15 cases with exclusive or near-exclusive subcutaneous involvement. The American Journal of Dermatopathology, 30(4), 327–332. 10.1097/DAD.0b013e31817d32b2
Bernard J., Poulalhon N., Argenziano G., Debarbieux S., Dalle S., Thomas L. (2013). Dermoscopy of dermatofibrosarcoma protuberans: A study of 15 cases. British Journal of Dermatology, 169(1), 85–90. 10.1111/bjd.12318
Bowne W. B., Antonescu C. R., Leung D. H., Katz S. C., Hawkins W. G., Woodruff J. M., Brennan M. F., Lewis J. J. (2000). Dermatofibrosarcoma protuberans: A clinicopathologic analysis of patients treated and followed at a single institution. Cancer, 88(12), 2711–2720.
Caccavale S., Martins Basso A., Vitiello P., Ronchi A., Sica A., Verolino P., Toncic R. J., Argenziano G. (2022). Dermatofibrosarcoma protuberans: Experience at a third-level referral center. Dermatology Practical & Conceptual, 12(1), e2022033. 10.5826/dpc.1201a33
Cai H., Wang Y., Wu J., Shi Y. (2012). Dermatofibrosarcoma protuberans: Clinical diagnoses and treatment results of 260 cases in China. Journal of Surgical Oncology, 105(2), 142–148. 10.1002/jso.22000
Connelly J. H., Evans H. L. (1992). Dermatofibrosarcoma protuberans. A clinicopathologic review with emphasis on fibrosarcomatous areas. American Journal of Surgical Pathology, 16(10), 921–925.
Dagan R., Morris C. G., Zlotecki R. A., Scarborough M. T., Mendenhall W. M. (2005). Radiotherapy in the treatment of dermatofibrosarcoma protuberans. American Journal of Clinical Oncology, 28(6), 537–539. 10.1097/01.coc.0000171278.69291.64
De Antoni E., Brambullo T., Pescarini E., Salmaso R., Bassetto F., Vindigni V. (2020). Dermatofibrosarcoma protuberans on tattooed skin: A case report. Advances in Skin & Wound Care, 33(2), 104–108. 10.1097/01.ASW.0000613548.11947.b4
Enneking W. F., Spanier S. S., Goodman M. A. (1980). A system for the surgical staging of musculoskeletal sarcoma. Clinical Orthopaedics and Related Research, (153), 106–120.
Foroozan M., Sei J. F., Amini M., Beauchet A., Saiag P. (2012). Efficacy of Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans: Systematic review. Archives of Dermatology, 148(9), 1055–1063. 10.1001/archdermatol.2012.1440
Gloster H. M. (1996). Dermatofibrosarcoma protuberans. Journal of the American Academy of Dermatology, 35(3, Pt 1), 355–374. 10.1016/s0190-9622(96)90597-6
Haas R. L., Keus R. B., Loftus B. M., Rutgers E. J., van Coevorden F., Bartelink H. (1997). The role of radiotherapy in the local management of dermatofibrosarcoma protuberans. Soft Tissue Tumours Working Group. European Journal of Cancer, 33(7), 1055–1060. 10.1016/s0959-8049(97)00026-9
Har-Shai Y., Govrin-Yehudain J., Ullmann Y., Kerner H., Cohen H. I., Lichtig C., Bergman R., Cohen A., Kuten A., Friedman-Birnbaum R. (1993). Dermatofibrosarcoma protuberans appearing during pregnancy. Annals of Plastic Surgery, 31(1), 91–93.
Haycox C. L., Odland P. B., Olbricht S. M., Piepkorn M. (1997). Immunohistochemical characterization of dermatofibrosarcoma protuberans with practical applications for diagnosis and treatment. Journal of the American Academy of Dermatology, 37(3, Pt 1), 438–444. 10.1016/s0190-9622(97)70146-4
Hirano S. A., Torosky C. M. (2012). Dermatofibrosarcoma protuberans arising at a Rho(D) immune globulin injection site. Cutis, 90(5), 233–234.
Kamino H., Reddy V. B., Gero M., Greco M. A. (1992). Dermatomyofibroma. A benign cutaneous, plaque-like proliferation of fibroblasts and myofibroblasts in young adults. Journal of Cutaneous Pathology, 19(2), 85–93. 10.1111/j.1600-0560.1992.tb01348.x
Kreicher K. L., Kurlander D. E., Gittleman H. R., Barnholtz-Sloan J. S., Bordeaux J. S. (2016). Incidence and survival of primary dermatofibrosarcoma protuberans in the United States. Dermatologic Surgery, 42(Suppl 1), S24–S31. 10.1097/DSS.0000000000000300
Lemm D., Mügge L. O., Mentzel T., Höffken K. (2009). Current treatment options in dermatofibrosarcoma protuberans. Journal of Cancer Research and Clinical Oncology, 135(5), 653–665. 10.1007/s00432-009-0550-3
Lindner N. J., Scarborough M. T., Powell G. J., Spanier S., Enneking W. F. (1999). Revision surgery in dermatofibrosarcoma protuberans of the trunk and extremities. European Journal of Surgical Oncology, 25(4), 392–397. 10.1053/ejso.1999.0663
Llombart B., Sanmartín O., López-Guerrero J. A., Monteagudo C., Serra C., Requena C., Poveda A., Vistós J. L., Almenar S., Llombart-Bosch A., Guillén C. (2009). Dermatofibrosarcoma protuberans: Clinical, pathological, and genetic (COL1A1-PDGFB) study with therapeutic implications. Histopathology, 54(7), 860–872. 10.1111/j.1365-2559.2009.03310.x
Llombart B., Serra-Guillén C., Monteagudo C., López Guerrero J. A., Sanmartín O. (2013). Dermatofibrosarcoma protuberans: A comprehensive review and update on diagnosis and management. Seminars in Diagnostic Pathology, 30(1), 13–28. 10.1053/j.semdp.2012.01.002
Maki R. G., Awan R. A., Dixon R. H., Jhanwar S., Antonescu C. R. (2002). Differential sensitivity to imatinib of 2 patients with metastatic sarcoma arising from dermatofibrosarcoma protuberans. International Journal of Cancer, 100(6), 623–626. 10.1002/ijc.10535
Martin E. C. S., Vyas K. S., Batbold S., Erwin P. J., Brewer J. D. (2022). Dermatofibrosarcoma protuberans recurrence after wide local excision versus Mohs micrographic surgery: A systematic review and meta-analysis. Dermatologic Surgery, 48(5), 479–485. 10.1097/DSS.0000000000003411
Mavili M. E., Gursu K. G., Gokoz A. (1994). Dermatofibrosarcoma with lymph node involvement. Annals of Plastic Surgery, 32(4), 438–440. 10.1097/00000637-199404000-00022
Mentzel T., Beham A., Katenkamp D., Dei Tos A. P., Fletcher C. D. (1998). Fibrosarcomatous ("high-grade") dermatofibrosarcoma protuberans: Clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. American Journal of Surgical Pathology, 22(5), 576–587. 10.1097/00000478-199805000-00009
Paradisi A., Abeni D., Rusciani A., Cigna E., Wolter M., Scuderi N., Kaufmann R., Podda M. (2008). Dermatofibrosarcoma protuberans: Wide local excision vs. Mohs micrographic surgery. Cancer Treatment Reviews, 34(8), 728–736. 10.1016/j.ctrv.2008.06.002
Parlette L. E., Smith C. K., Germain L. M., Rolfe C. A., Skelton H. (1999). Accelerated growth of dermatofibrosarcoma protuberans during pregnancy. Journal of the American Academy of Dermatology, 41(5, Pt 1), 778–783. 10.1016/s0190-9622(99)70023-x
Patel K. U., Szabo S. S., Hernandez V. S., Prieto V. G., Abruzzo L. V., Lazar A. J., López-Terrada D. (2008). Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Human Pathology, 39(2), 184–193. 10.1016/j.humpath.2007.06.009
Rouhani P., Fletcher C. D., Devesa S. S., Toro J. R. (2008). Cutaneous soft tissue sarcoma incidence patterns in the U.S.: An analysis of 12,114 cases. Cancer, 113(3), 616–627. 10.1002/cncr.23571
Salgado R., Llombart B., M Pujol R., Fernández-Serra A., Sanmartín O., Toll A., Rubio L., Segura S., Barranco C., Serra-Guillén C., Yébenes M., Salido M., Traves V., Monteagudo C., Sáez E., Hernández T., de Álava E., Llombart-Bosch A., Solé F., Guillén C., Espinet B., López-Guerrero J. A. (2011). Molecular diagnosis of dermatofibrosarcoma protuberans: A comparison between reverse transcriptase-polymerase chain reaction and fluorescence in situ hybridization methodologies. Genes Chromosomes & Cancer, 50(7), 510–517. 10.1002/gcc.20874
Sandberg A. A., Bridge J. A. (2003). Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors. Dermatofibrosarcoma protuberans and giant cell fibroblastoma. Cancer Genetics, 140(1), 1–12. 10.1016/s0165-4608(02)00848-8
Sanz-Trelles A., Ayala-Carbonero A., Rodrigo-Fernández I., Weil-Lara B. (1998). Leiomyomatous nodules and bundles of vascular origin in the fibrosarcomatous variant of dermatofibrosarcoma protuberans. Journal of Cutaneous Pathology, 25(1), 44–49. 10.1111/j.1600-0560.1998.tb01688.x
Shah A., Tassavor M., Sharma S., Torbeck R. (2021). The various treatment modalities of dermatofibrosarcoma protuberans. Dermatology Online Journal, 27(6). 10.5070/D327654070
Simon M. P., Pedeutour F., Sirvent N., Grosgeorge J., Minoletti F., Coindre J. M., Terrier-Lacombe M. J., Mandahl N., Craver R. D., Blin N., Sozzi G., Turc-Carel C., O'Brien K. P., Kedra D., Fransson I., Guilbaud C., Dumanski J. P. (1997). Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nature Genetics, 15(1), 95–98. 10.1038/ng0197-95
Sirvent N., Maire G., Pedeutour F. (2003). Genetics of dermatofibrosarcoma protuberans family of tumors: From ring chromosomes to tyrosine kinase inhibitor treatment. Genes Chromosomes & Cancer, 37(1), 1–19. 10.1002/gcc.10202
Sun L. M., Wang C. J., Huang C. C., Leung S. W., Chen H. C., Fang F. M., Huang E. Y., Lee S. P. (2000). Dermatofibrosarcoma protuberans: Treatment results of 35 cases. Radiotherapy and Oncology, 57(2), 175–181. 10.1016/s0167-8140(00)00228-0
Tanwar P., Singh A., Pratap S., Rattan A., Minhas S. S. (2021). Dermatofibrosarcoma—An uncommon entity, commonly mismanaged: A case report. Internation Journal of Surgical Case Reports, 87, 106385. 10.1016/j.ijscr.2021.106385
Zelger B., Sidoroff A., Stanzl U., Fritsch P. O., Ofner D., Jasani B., Schmid K. W. (1994). Deep penetrating dermatofibroma versus dermatofibrosarcoma protuberans. A clinicopathologic comparison. American Journal of Surgical Pathology, 18(7), 677–686. 10.1097/00000478-199407000-00003
留言 (0)