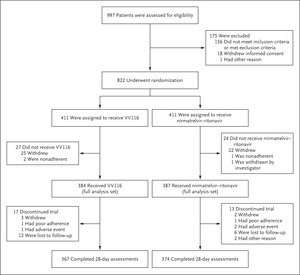
Trial Design and Randomization
In this multicenter, observer-blinded, randomized, controlled trial, symptomatic participants at high risk for progression to severe Covid-19 were randomly assigned in a 1:1 ratio to receive either oral VV116 (600 mg every 12 hours on day 1 and 300 mg every 12 hours on days 2 through 5) or oral nirmatrelvir–ritonavir (300 mg of nirmatrelvir plus 100 mg of ritonavir every 12 hours for 5 days) (Fig. S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org). VV116 was manufactured and provided by Vigonvita Life Sciences. The selection of nirmatrelvir–ritonavir as the active control for comparison with VV116 was based on the established superiority of nirmatrelvir–ritonavir to placebo12 and on its recommendation as the standard treatment for our target population by the WHO guideline.11
Randomization was performed with the use of a centralized, interactive Web response system. All the site investigators, site staff (except for those who administered the trial drugs), and those who were involved in end-point assessments were unaware of the trial-group assignments until unblinding on May 20, 2022. Participants remained aware of the trial-group assignments throughout the trial. Additional details are provided in the Supplementary Appendix.
The data-cutoff date for the primary analysis was May 13, 2022, when the target number of primary end-point events (>724 events) was reached in the full analysis population. The data-cutoff date for the final analysis was August 18, 2022.
Trial Oversight
The trial was approved by the National Human Genetic Resources Committee in China and the institutional review board or ethics committee at each trial site before the start of recruitment and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. One of the sponsors, Vigonvita Life Sciences, designed and monitored the trial and collected and analyzed the data in collaboration with the site investigators. Safety oversight was performed by Vigonvita Life Sciences and the institutional review board or ethics committee at each site. The first author drafted the manuscript, and the writing committee revised the manuscript and made the decision to submit it for publication. All the authors had data confidentiality agreements with Vigonvita Life Sciences and vouch for the accuracy and completeness of the data and for the fidelity of the trial to the protocol, available at NEJM.org.
Participants
After written informed consent was obtained, participants from seven hospitals in Shanghai, China, that were designated by the Chinese government for the treatment of Covid-19 were assessed for eligibility between April 4, 2022, and May 2, 2022. Adults 18 years of age or older were eligible if they had mild-to-moderate Covid-19 with a total symptom score of 2 or more as determined on the basis of definitions adapted from the Food and Drug Administration.18 Symptom scores range from 0 to 3 (with higher scores indicating greater severity) for each of 11 symptoms; total symptom scores range from 0 to 33 (Table S1). Other key inclusion criteria were a positive SARS-CoV-2 reverse-transcriptase–polymerase-chain-reaction (RT-PCR) test with an additional finding indicating early infection or high viral activity (the findings are listed in the Supplementary Appendix), and at least one risk factor for progression to severe Covid-19.
Key exclusion criteria were confirmed or suspected severe or critical Covid-19 or an anticipated need for mechanical ventilation before randomization, an alanine aminotransferase or aspartate aminotransferase level that was more than 1.5 times the upper limit of the normal range, an estimated glomerular filtration rate (eGFR) of less than 60 ml per minute, or the use of contraindicated drugs listed in the package insert of nirmatrelvir–ritonavir. Although nirmatrelvir–ritonavir is not contradicted in persons with an eGFR of 30 to less than 60 ml per minute, we excluded these participants to avoid a potential overdose in the updated protocol (version 3.0; April 10, 2022). Before that date, a total of 38 participants with an eGFR of 30 to less than 60 ml per minute had been enrolled in the trial (16 in the VV116 group and 22 in the nirmatrelvir–ritonavir group). Full eligibility criteria are provided in the Supplementary Appendix and protocol.
Assessment
Covid-19–related symptom scores (described above) and scores on the WHO Clinical Progression Scale (range, 0 to 10, with higher scores indicating a worse clinical condition) (Table S2) were determined by investigators on day 1 before the trial-drug administration, followed by assessment at approximately the same time every day until the resolution of Covid-19–related target symptoms or day 28, whichever was earlier. SARS-CoV-2 RNA from nasopharyngeal swabs was measured by RT-PCR assay at each site, with both qualitative data (positive or negative) and quantitative data (cycle-threshold value) obtained if available. More details of assessment and data collection are provided in the protocol.
End Points
The primary efficacy end point was the time from randomization to sustained clinical recovery through day 28. Sustained clinical recovery was defined as the alleviation of all Covid-19–related target symptoms to a total symptom score of 0 or 1 (range, 0 to 33, with higher scores indicating greater severity) for 2 consecutive days. The first day of the 2-consecutive-day period was considered to be the event date. Secondary efficacy end points included progression to severe or critical Covid-19 or death from any cause; the change in Covid-19–related symptom score and the score on the WHO Clinical Progression Scale through day 28, the time to sustained resolution of all target symptoms and to a first negative SARS-CoV-2 test, and clinical recovery, symptom resolution, and a negative SARS-CoV-2 test by prespecified days. Safety end points included adverse events and serious adverse events, with severity determined according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 5.0. Any adverse event that emerged or worsened from the time of informed consent through day 28 was actively recorded and reported for trial-regimen recipients. Details of the end points are provided in the Supplementary Appendix and Table S3. The primary end point was assessed in the primary analysis (data-cutoff date, May 13, 2022), and the data were updated in the final analysis (data-cutoff date, August 18, 2022).
Statistical Analysis
The primary efficacy hypothesis was that VV116 would be noninferior to nirmatrelvir–ritonavir with respect to sustained clinical recovery. Owing to the lack of data on the time to clinical recovery in participants with omicron infection treated with nirmatrelvir–ritonavir, the reference duration of 5.5 days was estimated on the basis of the duration of acute symptoms in persons infected with SARS-CoV-2 during the omicron wave19 and an overall vaccination rate of more than 90% in the general population in Shanghai.20 To satisfy the noninferiority hypothesis, the lower boundary of the two-sided 95% confidence interval for the hazard ratio of the primary end point had to be above 0.8. The noninferiority margin corresponds to a duration of 6.875 days to sustained clinical recovery, which is 25% longer than 5.5 days. A minimum of 724 events were required to ensure a statistical power of 85%.
The noninferiority hypothesis was tested in the full analysis population — that is, the modified intention-to-treat population (all the participants who underwent randomization and received at least one dose of VV116 or nirmatrelvir–ritonavir). Sensitivity analyses involved participants who started a trial regimen within 5 days after symptom onset and the per-protocol population. The intention-to-treat population (all the participants who underwent randomization) was analyzed post hoc. Details of the analysis populations are provided in Tables S4 and S5.
For all the other efficacy analyses, data were analyzed in the full analysis population. The Kaplan–Meier method was used to estimate the median time to sustained clinical recovery, with the 95% confidence interval estimated by means of the Brookmeyer–Crowley method with log–log transformation. The hazard ratio for time to sustained clinical recovery and its 95% confidence interval were estimated with the use of the Cox proportional-hazards model. Data for participants without sustained clinical recovery were censored on the last day on which Covid-19–related symptoms or signs were recorded. Participants with missing end-point data were considered to have not had clinical recovery on that day, and a sensitivity analysis was performed with the use of the multiple-imputation method. Subgroup analyses of the primary end point were prespecified to assess the consistency of the intervention effect. For efficacy results other than the primary end point in the full analysis population, 95% confidence intervals have not been adjusted for multiplicity and should not be used to infer treatment effects. Additional details are provided in the statistical analysis plan, available with the protocol.







留言 (0)