
Remember me
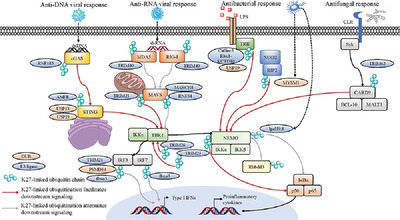
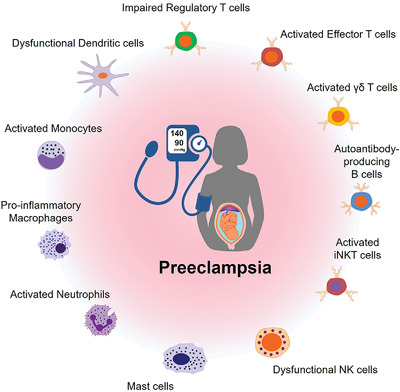
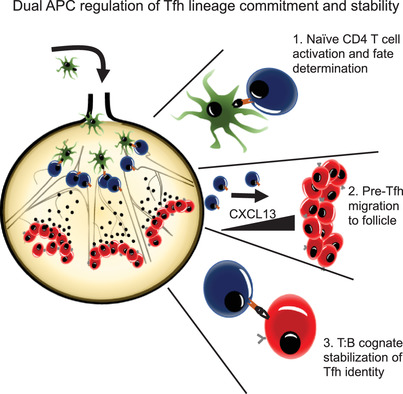
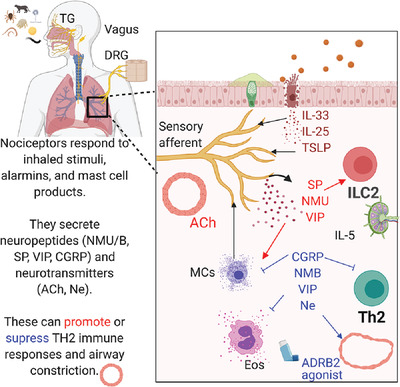
As an immunological response to inflammation, leukocytes are attracted to inflamed tissues by chemokines such as CC-chemokine ligand 2 (CCL2, a.k.a. MCP-1) and C-X-C motif chemokine ligand 2 (CXCL2, a.k.a. MIP-2).1 However, to avoid an excessive response, leukocyte infiltration should be halted for the resolution of inflammation, but not all the mechanisms that govern this are known.2 Here, we addressed the potential chemotactic effect of the chromogranin A (CgA)-derived peptide catestatin (CST: hCgA352-372).3, 4
CgA and its bioactive peptide CST are produced by endocrine cells, for example, chromaffin and pancreatic beta cells, and bone-marrow-derived cells such as macrophages (Mϕs).5-7 While the concentrations of CST in circulation are in the low nM range, concentrations in the μM range were detected in mouse tissues.3, 8-10 These much higher concentrations indicate that CST exerts its effects locally. CST has primarily anti-inflammatory properties, as CST reduces inflammation in cardiac and chronic inflammatory diseases and CST-knockout mice display increased inflammation in the intestine, heart, and adrenal gland.7, 8, 11, 12 However, CST has also pro-inflammatory effects as it is chemotactic11, 13, 14 and promotes the production of inflammatory chemokines (IL-8, CCL2-4).14 Nevertheless, administration of exogenous CST reduces monocyte and Mϕ infiltration in the liver, heart, and gut in mouse models of type II diabetes, hypertension, atherosclerosis, and colitis.7, 8, 11, 12, 15 In a colitis model, CST also reduced granulocyte infiltration in the colon.15 Finally, the adrenal gland, heart, and gut of CST knockout mice display increased Mϕ infiltration.7, 8, 11, 16 These findings raise the question of how CST affects leukocyte chemotaxis.
In this study, we show that while CST itself is chemotactic, it blocks the extravasation and migration of phagocytes both in vitro and in vivo. These findings support a model where the anti-inflammatory effects of CST could be partly the result of redirecting monocytes and neutrophils away from the inflammation sites.
2 RESULTS & DISCUSSIONTranswell migration assays on human blood monocytes showed only a weak chemotactic effect of CST in a broad concentration range (1 nM–5 μM) (Supplementary Fig. S1). In line with this, human blood monocytes migrated towards a high concentration of CST (5 μM) in the gradientech assay, but this migration was not significant and less efficient compared to the canonical inflammatory chemokine CCL2 (0.5 nM) (Fig. 1A-C). To investigate the effect of CST on phagocytes in vivo, we performed imaging of the mouse cremaster muscle to follow the rolling of monocytes (unstained) and neutrophils (Ly6G+ stained cells using intravenous injection of fluorescently labeled antibody against Ly6G) (Fig. 1D).14 Upon superfusion of the muscle with CST (5 μM), both monocytes and neutrophils decreased their speed and attached to the vessel wall with similar efficiency as with the inflammatory chemotactic agent CXCL2 (0.5 nM) (Fig. 1D-F; Supplementary Fig. S2A-D). Thus, both our in vivo and in vitro migration assays support that CST is chemotactic, although the effects in vivo in mice are stronger than for human monocytes in vitro. Possibly, the local surroundings in the tissues (e.g., epithelial cells or cytokines) enhance the chemotactic effect of CST in vivo. These findings raise the question of how CST can reduce the reported monocyte and granulocyte infiltration in inflamed tissues such as the liver (in diet-induced obese mice), intestine (in colitis model), heart (in hypertension model), and atheromatous plaques (in atherosclerosis model).7, 8, 11, 12, 15

CST is weakly chemotactic. (A) Scheme showing set-up of Gradientech migration assay. Two syringes filled with buffer ± chemoattractant were connected to the device (green) to create a flow (y-direction) and perpendicular (x) cytokine gradient. The inset shows the migration of monocytes along with the flow and towards the chemoattractant. (B) Representative tracks of human monocytes showing the x- and y-movement of individual cells upon exposure to the indicated buffer, 5 μM CST or 0.5 nM CCL2. (C) Quantification of panel B (N = 3). (D) Scheme showing set-up of cremaster muscle imaging in mice to visualize phagocyte (monocytes and neutrophils) extravasation in vivo. (E) Phagocyte rolling velocity (top) and attachment (bottom) upon overflowing the muscle with buffer (control, gray), 0.5 nM MIP-2 (blue) or 5 μM CST (black) for 90 minutes (imaged at T = 0, 30, 60, 90) (N = 3, two-way ANOVA). (F) Representative images of granulocyte attachment over time (imaged at T = 0, 30, 60, 90 minutes) to the vessel wall upon only buffer, CXCL2, or CST stimulation as visualized with an antibody against Ly6G (green). *P < 0.05; **P < 0.01; ***P < 0.001; ns: not significant
To address how CST affects Mϕ chemotaxis to inflamed tissues, we used the aortic ring vessel model17 (Fig. 2A), which is based on the co-embedding of part of the aorta of Cx3cr1+/gfp transgenic mice adjacent to isolated pancreatic islets.18 These islets secrete chemokines, such as vascular endothelial growth factor (VEGF)-A, resulting in the directional Mϕ migration from the aortic ring as well as vessel growth towards the pancreatic islets. Migration of CX3CR1+GFP Mϕs from the aortic ring was visualized by fluorescence microscopy19 (Fig. 2B-C; Supplementary Fig. S3). As expected, the CX3CR1-Mϕs moved toward the pancreatic islets in absence of CST (Figure 2B). However, perfusing the aortic ring with CST (5 μM) resulted in a lower number of CX3CR1+GFP Mϕs migrating toward the pancreatic islets (Figure 2B), indicating that CST blocked directional migration. Interestingly, we also observed that CST is pro-angiogenic, as it increased both the amount and length of the sprouts and branches emanating from the aortic rings (Fig. 2C-D; Fig. S4).
 CST blocks migration in the aortic ring model promotes angiogenesis. (A) Scheme showing set-up of aortic ring assay. Aortic ring was isolated from CX3CR1-GFP mice and embedded adjacent to pancreatic islets in collagen I. Image shows islets (blue), CD31 (red) and CX3CR1 (green). (B) Representative images of CX3CR1-Mϕ migration upon control or CST stimulation of the aortic ring. The graph shows the percentage of cells above (yellow) the center of mass (N = 8). (C) Representative images of vessels by CD31 labeling (red) upon control or CST stimulation of the aortic ring. (D) Quantification of angiogenesis (illustrated in Fig. S4A). Total number of sprouts and branches (left) and their length (right) (N = 5-6, Mann-Whitney test; *P P
CST blocks migration in the aortic ring model promotes angiogenesis. (A) Scheme showing set-up of aortic ring assay. Aortic ring was isolated from CX3CR1-GFP mice and embedded adjacent to pancreatic islets in collagen I. Image shows islets (blue), CD31 (red) and CX3CR1 (green). (B) Representative images of CX3CR1-Mϕ migration upon control or CST stimulation of the aortic ring. The graph shows the percentage of cells above (yellow) the center of mass (N = 8). (C) Representative images of vessels by CD31 labeling (red) upon control or CST stimulation of the aortic ring. (D) Quantification of angiogenesis (illustrated in Fig. S4A). Total number of sprouts and branches (left) and their length (right) (N = 5-6, Mann-Whitney test; *P P
The loss of directional cell migration to the pancreatic islets might be caused by blockage of chemokine-induced cell migration by CST. To investigate this possibility, we performed intravital imaging of the cremaster muscle, but this time for CST in combination with CXCL2. This resulted in the inverse effect compared to CST or CXCL2 alone: release of attached cells from the vessel wall and reduced migration of cells into the tissue (Fig. 3A; Supplementary Fig. S5A-C), indicating that despite being chemotactic, CST blocks CXCL2 elicited phagocyte recruitment. To confirm that CST exerts this effect on both monocytes and neutrophils, we performed intravital imaging of the cremaster muscle with intravenous injection of CD115+ antibody to stain monocytes and Ly6G+ antibody to stain neutrophils. Again, the combination of CST and CXCL2 resulted in less recruitment of both monocytes (CD115+) and neutrophils (Ly6G+) compared to CST or CXCL2 alone (Fig. 3B-C).

CST blocks migration induced by inflammatory cytokines. (A) Cremaster muscle imaging. Phagocyte attachment to vessel wall upon overflowing the muscle with buffer (control, gray) and buffer with the chemoattractant CXCL2 (blue), CST (black), or both (red) (N = 3, two-way ANOVA). (B) Representative images of granulocyte and monocyte attachment at T = 90 min to the vessel wall upon only buffer, CXCL2, CST, or stimulation with both, as visualized with antibodies against Ly6G (green) and CD115 (red). (C) Monocyte (upper) and granulocyte (lower) attachment to vessel wall upon overflowing the muscle with buffer (control, gray) and buffer with the chemoattractant CXCL2 (blue), CST (black), or both (red). (D) Representative hematoxylin-eosin (HE) images of mouse phagocyte migration in transwell assay toward IL-8, CST, or both. (E) Graphs displaying migrated mouse neutrophils (left) and monocytes (right) toward the lower compartment of the transwell assay filled with only medium or medium containing IL-8, CST, or both. (F) Percentages of migrated human monocytes toward the lower compartment of the transwell assay filled with only medium or medium containing CCL2, CST, or both. (G) Gradientech migration assay. Representative x- and y-movement of human monocytes exposed to opposite gradients of CST and CCL2 (N = 3). Mann-Whitney test *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns: not significant
To test if CST could also block chemotaxis to other chemokines, we performed in vitro transwell migration assays with both human and mouse phagocytes (Fig. 3; Supplementary Fig. S6). While IL-8 alone attracted mouse neutrophils as expected,20 IL-8 in combination with CST blocked the neutrophil migration to the lower compartment of the transwell (Fig. 3D and E). The combination of IL-8 and CST did not affect the migration of human monocytes (Supplementary Fig. S6D). CST did not block migration of mouse phagocytes and human monocytes towards the chemoattractive peptide N-formyl-methionyl-leucyl-phenylalanine (fMLP) (Supplementary Fig. S6A-C).
Another factor that can influence immune cell migration is their exposure to microbial agents, such as lipopolysaccharide (LPS), which induce their activation.21 To determine whether pre-treatment of the cells with LPS or CST can influence the blocking effect of CST, we pre-stimulated human monocytes 30 minutes with LPS or CST before performing the in vitro transwell migration assays (Supplementary Fig. S7). Again, we observed that CST could block CCL2-induced monocyte migration, and, although not significant, this effect did not seem altered by pretreatment of the cells with LPS or CST (Fig. 3F; Supplementary Fig. S7B). To confirm the observed block of chemotaxis in human monocytes by CST, we performed an in vitro gradientech migration assay with a gradient of CCL2 in presence of CST (Fig. 3G). Similar to our findings with the intravital imaging, CST blocked monocyte migration toward CCL2.
Our findings indicate that CST counteracts the chemoattraction of leukocytes by inflammatory chemokines CCL2, CXCL2, and IL-8. This could contribute to the previously observed reduced immune infiltrate in the liver, heart, and gut in mouse models of type II diabetes, hypertension, atherosclerosis, and colitis upon treatment with CST.7, 8, 11, 12, 15 However, CST has been reported to promote the production of chemokines14 and we confirmed previous findings that CST is chemotactic itself,11, 13, 14 indicating that the regulation of chemotaxis by CST is complex. To understand this regulation, the receptor(s) for CST needs to be identified. We speculate that this might be a G-protein coupled receptor (GPCR), since GPCRs are actively involved in leukocyte migration22 and are widely expressed in all cell types responsive to CST (e.g., monocytes,13 neutrophils,23, 24 Mϕs,7, 8, 11, 12, 15 endothelial,12, 25 and mast cells).14 Moreover, CCL2, CXCL2, and IL-8 all signal via GPCRs.26 Possibly, CST treatment causes a heterologous desensitization to these chemoattractants.
Another open question is how the chemotactic properties of CST are regulated in vivo. CgA, the precursor of CST, is co-released with catecholamines by neuroendocrine cells4, 27-29 suggesting that these cells might prevent or limit immune cell infiltration in an activity-based fashion. However, neutrophils and Mϕs also produce CST themselves,7, 23, 30, 31 arguing that CST might downregulate their infiltration in an autocrine fashion.
In any case, our data suggest that CST reduces the infiltration of monocytes and Mϕs in inflamed tissues,7, 8, 11, 12, 15 offering a possible mechanistic explanation for the correlation of CST levels with improved disease outcome in patients suffering from chronic diseases.8-11, 32 These findings reinforce the emerging concept that CST could be a therapeutic target for the treatment of diseases associated with chronic inflammation.3, 7, 8, 11, 33
AUTHORSHIPE.M.M., G.C., S.K.M., and G.v.d.B. designed the study. E.M.M, K.P., and G.C. designed and performed the experiments. N.K.R. contributed to the in vitro mice experiments. E.M.M. and G.v.d.B wrote the manuscript and all authors (E.M.M., K.P., G.C., M.P., S.K.M., and G.v.d.B) participated in discussing and editing the manuscript.
DISCLOSUREThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
FUNDINGG.v.d.B. is funded by a Young Investigator Grant from the Human Frontier Science Program (HFSP; RGY0080/2018), and a Vidi grant from the Netherlands Organization for Scientific Research (NWO-ALW VIDI 864.14.001). G.v.d.B. has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No. 862137. S.K.M. is supported by a grant from the US Department of Veterans Affairs (I01BX000323). G.C. is supported by grants from the Swedish Research Council, the Swedish Society for Medical Research, and the Göran Gustafsson foundation. E.M.M. is supported by a short-term EMBO fellowship (EMBO7887).
Comments (0)