
記住我


MED12 is a subunit in a kinase module that plays a key role in cell differentiation and development through RNA-transcription.1, 2 The module regulates a larger protein complex known as the Mediator, encoded by 26 human genes, that facilitates developmental signalling from DNA-bound transcription factors to RNA polymerase II.2, 3 Missense variants in MED12 are associated with X-linked recessive and clinically overlapping developmental disorders including Lujan-Fryns syndrome (OMIM 309520), Ohdo syndrome4 (OMIM 300895) and Opits-Kaveggia syndrome (OMIM 305450), which all, inter alia, are characterized by intellectual disability and facial dysmorphism in affected males.5, 6 However, a missense variant p.(Arg1148His) in MED12 was recently reported in two affected sisters with intellectual disability, macrocephaly and deep set eyes,7 which challenges the original view of MED12-related disorders as being classically X-linked recessive. Interestingly, the X-chromosome inactivation profile in blood cells from the two sisters showed no correlation with clinical outcomes as previously reported in symptomatic female carriers of an X-linked disease.8 Likewise, an intronic splice variant causing exon 9 skipping in MED12 was recently reported in a girl with generalized hypotonia, cleft lip and palate, facial dysmorphism and pulmonary stenosis.9 The clinical spectrum of MED12-related disorders was further expanded when de novo loss of function variants in MED12 was shown to be associated with Hardikar syndrome (OMIM 612726) in females.10 The affected patients had facial cleft, pigmentary retinopathy, aortic coarctation, biliary anomalies and intestinal malrotation. Finally, female mice that are heterozygous for a conditional null allele of Med12 expressed in a mosaic fashion die prenatally and show split face exencephaly, spina bifida and craniorachishisis,11 which further substantiates the essential role of MED12 in female embryonic development. In this report, we describe, to our knowledge, the first case of a female fetus with severe malformations and heterozygous for a MED12 de novo nonsense variant. Understanding the degree to which MED12 genetic heterogeneity affects human female fetuses is vital for genetic counselling of couples when MED12-related disorders are diagnosed prenatally.
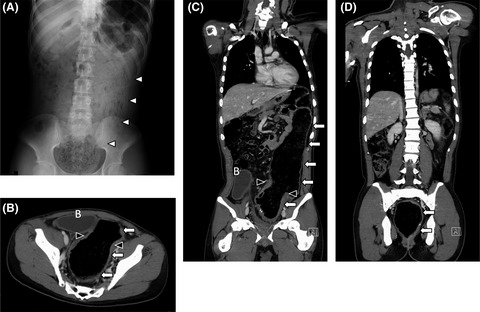
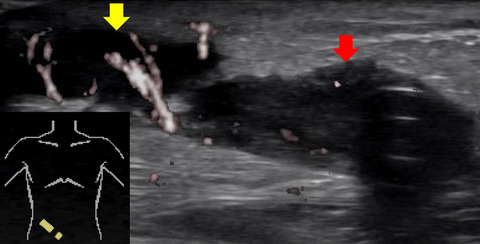
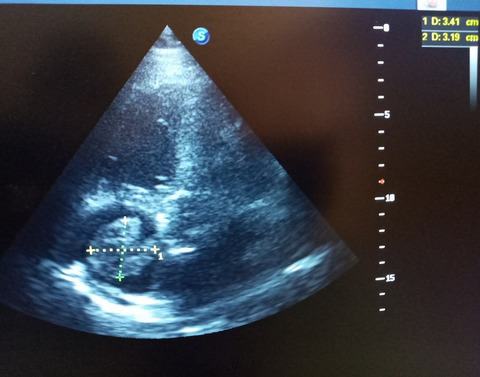
2 CASE PRESENTATIONA non-consanguineous couple, with two healthy sons, presented in their third pregnancy. The result of first trimester risk assessment was normal and no ultrasound anomalies were seen at gestational age (GA) 13+4. At GA 14+5, an ultrasound scan was performed suggesting cleft lip and cleft palate. At GA 16+0, severe bilateral cleft lip and palate was confirmed and right-sided aortic arch detected. Array-CGH (SurePrint G3 Human CGH 4x180K arrays, Agilent Technologies) of a chorionic villus sample revealed a normal female karyotype. A subsequent scan at 16+5 weeks additionally identified hypertelorism (interorbital distance of +2SD), microphthalmia, double outlet right ventricle and a small dorsally displaced gastric ventricle as shown in Figure 1A–E. After genetic counselling, the pregnancy was terminated at 17+6 weeks on parental request. Fetal autopsy showed a midline facial malformation with bilateral cleft lip and cleft palate and anterior displacement of the premaxilla, as well as ventricular septal defect. Aplasia of the diaphragm caused displacement of the liver into the thoracic cavity explaining pulmonary hypoplasia (Figure 1F). The appendix and cecum were displaced to the left part of the abdominal cavity indicating partial malrotation of the intestinal tract. The uterus, ovaries and vagina were not identifiable. Full body X-ray showed no signs of skeletal abnormalities.

(A) Bilateral cleft lip and cleft palate. (B) Dysmorphism of the nose. (C) Hypertelorism. (D) Ventricular septal defect. (E) Gastric ventricle is located behind the heart (red arrow). (F) Autopsy of abdomen showing displacement of the liver into the thorax due to diaphragm aplasia
3 METHODS AND RESULTSThe procedures described in this report involving patient data are in agreement with the ethical standards of Aarhus University Hospital, Denmark. Informed consent was obtained prior to all procedures, as well as for publication.
At week 17+3, DNA was isolated from cells obtained by amniocentesis. QF-PCR analysis showed no signs of maternal contamination. Maternal and paternal DNA derived from blood was used for trio-exome sequencing. Fetal, maternal and paternal DNA was sequenced using NextSeq 500 (Illumina) and NimbleGen MedExome (Roche) with an average coverage of >94x. The reads were processed using BWA-MEM 0.7.5a and GATK version 3.6. Data analysis was conducted at the Department of Clinical Genetics, Aarhus University Hospital, using VarSeq analysis software 2.2.2 (Golden Helix) and reference genome hg19. Variants were filtered based on inheritance patterns (autosomal dominant de novo, autosomal recessive; homozygous/compound heterozygous).
The analysis identified heterozygosity for a de novo variant in MED12 NM_005120.3: c.6196C>T (p.(Gln2066*)) (Figure 2). This variant is not in the gnomAD database (v2.1.1), nor has it previously been associated with disease. The variant has been uploaded to HGMD 2021.1 as part of a previous report assessing the clinical utility of fetal exomes.12 As the MED12 variant is predicted to introduce a premature stop codon into exon 42 of the transcript that contains 45 exons in total, it is likely that the transcript has undergone nonsense-mediated decay.13 Combined Annotation-Dependent Depletion (CADD) assessment of variant deleteriousness scored the variant to 37, suggesting a pathogenic effect.14 A full list of detected variants is provided in Table S1.

Pedigree of index family. Squares indicate healthy males. Circles indicate females. Black triangle indicates termination of pregnancy of the female fetus. Sanger sequencing is shown for the parents and the female fetus demonstrating heterozygosity for a de novo nonsense variant in MED12 c.6196C>T (p.(Gln2066*))
4 DISCUSSIONTo our knowledge, this is the first report of a de novo MED12 nonsense variant in prenatal diagnosis of fetal anomalies. The identified variant may cause MED12 deficiency, and we suggest that this explains the bilateral cleft lip and palate, micropthalmia, hypertelorism, cardiac malformations, diaphragm agenesis and intestinal malrotation in the female fetus reported here. The observed prenatal phenotype is partly overlapping with the few cases of postnatal MED12-related disorders in females.9, 10 Specifically, facial cleft, cardiac anomalies and intestinal malrotation have previously been observed in the few postnatal cases of females with Hardikar syndrome due to MED12 nonsense variants.10 The split face that has been observed in female mice11 fetuses carrying a conditional heterozygous Med12 null allele bears striking resemblance to the facial dysmorphism observed in this human fetus. However, we did not find any evidence of exencephaly or spina bifida, which might be explained by species-specific effects of MED12 deficiency. Also, to our knowledge, diaphragm agenesis has not previously been reported in MED12-related disorders, which might constitute a novel phenotypic marker related to MED12 deficiency. This phenotypic diversity may alternatively be related to tissue specific X-inactivation, but has not been examined here due to nonavailability of fetal tissue.15, 16 Further studies of MED12 variants in prenatal diagnostics are warranted to understand and substantiate our findings, which will be instrumental in genetic counselling of MED12-related disorders.
ACKNOWLEDGEMENTSWe thank the affected family for their participation in this report.
CONFLICTS OF INTERESTSThe authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONSLF gathered the data and drafted the manuscript. NB and LA performed the NGS analysis and helped to finalize the manuscript. MC conducted the ultrasound scans and helped to finalize the manuscript. LF made the autopsy and helped to finalize the manuscript. IV conceptualized the study and helped to finalize the manuscript.
ETHICAL APPROVALThis report was conducted in accordance with the World Medical Association Declaration of Helsinki of 1964 and local principles for medical research of Aarhus University Hospital, Denmark.
CONSENTThe affected family provided informed consent for the participation and publication of this report.
The data that supports the findings of this study are available in the supplementary material of this article.
Filename Description ccr35124-sup-0001-TabS1.xlsxapplication/excel, 60.5 KB Tab S1Please note: The publisher is not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing content) should be directed to the corresponding author for the article.
REFERENCES
1Rocha PP, Scholze M, Bleiss W, Schrewe H. Med12 is essential for early mouse development and for canonical Wnt and Wnt/PCP signaling. Development. 2010; 137: 2723- 2731. doi:10.1242/dev.053660 2Calpena E, Hervieu A, Kaserer T, et al. De novo missense substitutions in the gene encoding CDK8, a regulator of the mediator complex, cause a syndromic developmental disorder. Am J Hum Genet. 2019; 104: 709- 720. doi:10.1016/j.ajhg.2019.02.006 3Harper TM, Taatjes DJ. The complex structure and function of mediator. J Biol Chem. 2018; 293: 13778- 13785. doi:10.1074/jbc.R117.794438 4Murakami H, Enomoto Y, Tsurusaki Y, Sugio Y, Kurosawa K. A female patient with X-linked Ohdo syndrome of the Maat-Kievit-Brunner phenotype caused by a novel variant of MED12. Congenit Anom. 2020; 60: 91- 93. doi:10.1111/cga.12350 5Graham JM Jr, Schwartz CE. MED12 related disorders. Am J Med Genet A. 2013; 161a: 2734- 2740. doi:10.1002/ajmg.a.36183 6Popp B, Ekici AB, Thiel CT, et al. Exome pool-Seq in neurodevelopmental disorders. Eur J Hum Genet. 2017; 25: 1364- 1376. doi:10.1038/s41431-017-0022-1 7Charzewska A, Maiwald R, Kahrizi K, et al. The power of the mediator complex-expanding the genetic architecture and phenotypic spectrum of MED12-related disorders. Clin Genet. 2018; 94: 450- 456. doi:10.1111/cge.13412 8Lesca G, Moizard MP, Bussy G, et al. Clinical and neurocognitive characterization of a family with a novel MED12 gene frameshift mutation. Am J Med Genet A. 2013; 161a: 3063- 3071. doi:10.1002/ajmg.a.36162 9Wang C, Lin L, Xue Y, et al. MED12-related disease in a Chinese girl: clinical characteristics and underlying mechanism. Front Genet. 2020; 11: 129. doi:10.3389/fgene.2020.00129 10Li D, Strong A, Shen KM, et al. De novo loss-of-function variants in X-linked MED12 are associated with Hardikar syndrome in females. Genet Med. 2020; 23(4): 637- 644. doi:10.1038/s41436-020-01031-7 11Rocha PP, Bleiss W, Schrewe H. Mosaic expression of Med12 in female mice leads to exencephaly, spina bifida, and craniorachischisis. Birth Defects Res A Clin Mol Teratol. 2010; 88: 626- 632. doi:10.1002/bdra.20693 12Becher N, Andreasen L, Sandager P, et al. Implementation of exome sequencing in fetal diagnostics-data and experiences from a tertiary center in Denmark. Acta Obstet Gynecol Scand. 2020; 99: 783- 790. doi:10.1111/aogs.13871 13Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol. 2015; 16: 665- 677. doi:10.1038/nrm4063 14Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014; 46: 310- 315. doi:10.1038/ng.2892 15Tukiainen T, Villani AC, Yen A, et al. Landscape of X chromosome inactivation across human tissues. Nature. 2017; 550: 244- 248. doi:10.1038/nature24265 16Shvetsova E, Sofronova A, Monajemi R, et al. Skewed X-inactivation is common in the general female population. Eur J Hum Genet. 2019; 27: 455- 465. doi:10.1038/s41431-018-0291-3
留言 (0)