
Remember me
Noradrenaline is a major monoaminergic neurotransmitter and has many important functions through action on adrenergic receptors, such as α1, α2, and β receptors.1 It has known that noradrenaline is also involved in pain modulation. Among three adrenergic receptors, α2-adrenergic receptors (α2-ARs) play a key role in mediating pain modulatory effects of noradrenaline.2, 3 The α2-ARs are distributed in the pain signaling pathway, including primary afferents and spinal dorsal horn.4-7 In the spinal cord, norepinephrine released from descending pathways results in a presynaptic inhibition of pain by activating α2-ARs on central terminals of primary afferent nociceptors.2 The α2-AR agonists can also mimic the noradrenergic projection of descending pain inhibition.2 Dexmedetomidine (DEX), a potent highly selective α2A-AR agonist, has shown potential analgesic effects in animals and humans when administered intrathecally or systemically.8-10 The α2 adrenergic drugs that include DEX and clonidine are approved as analgesic agents in clinical settings.11
Systemic DEX analgesia could be blocked by peripheral α2-AR antagonists in neuropathic pain, suggesting a peripheral anti-nociceptive effect of DEX.12, 13 The peripheral effect of DEX on nociception is mediated by peripheral α2-ARs, which have been identified in the dorsal root ganglion (DRG). Mechanisms of DEX peripheral analgesia may underlie the modulation of a number of ligand-gated and voltage-gated ion channels, which are also expressed in DRG neurons. For example, DEX has been found to inhibit sodium channels through α2-ARs located on DRG and the trigeminal ganglion neurons.14, 15 DEX also suppresses the activity of TRPV1 via an α2-ARs and cAMP / protein kinase A (PKA) signaling pathway in DRG neurons.16
P2X3 receptor is a purinergic ATP receptor. P2X3 homomeric and P2X2/3 heteromer receptors are distributed in DRG neurons and mainly located in a subset of small and medium-sized nociceptive neurons.17-19 These peripheral P2X3-containing receptors contribute to the transmission of nociceptive signaling. For example, blocking P2X3 receptors by antagonists or antisense oligonucleotide can effectively reduce nociception.20, 21 P2X3 receptor-mediated currents in DRG neurons and nociceptive behaviors increase after inflammation and nerve injury.22-25 It has shown that P2X3 receptors are regulated by adrenergic signaling. The mRNA assessments indicate both α1-ARs and α2-ARs are expressed in DRG.3, 4 Noradrenaline potentiates ATP-evoked currents in DRG neurons by activating PKC via Gq protein-coupled α1-ARs.26 Considering the presence of α2-ARs and P2X3 receptors in DRG neurons, it was still unclear whether P2X3 receptors were also modulated by activation of α2-ARs. Herein, we observed that the activation of α2A-ARs by DEX inhibited the electrophysiological activity of P2X3 receptors via an intracellular cAMP signaling pathway in rat DRG neurons. DEX also relieved P2X3 receptor-mediated nociceptive behaviors in rats by activating peripheral α2A-ARs.
2 MATERIALS AND METHODS 2.1 Preparation of DRG neuronsAll experimental protocols were approved by the animal research ethics committee of Hubei University of Science and Technology. All animal data reporting has followed the ARRIVE 2.0 guidelines (PMID: 32663096). Sprague-Dawley male rats (5–6 weeks old) were anesthetized and then killed. The DRGs from rats were removed and chopped. The minced ganglia were transferred to a test tube containing Dulbecco's modified Eagle's medium (DMEM) and incubated in a shaking for 25–30 min at 35°C. Incubation solution contained 1.0 mg/ml collagenase, 0.5 mg/ml trypsin, and 0.1 mg/mL IV DNase. Trypsin digestion was terminated by adding 1.25 mg/mL soybean trypsin inhibitor. The cells were cultured for 12–24 hours at 37°C in DMEM containing never growth factor (100 ng/mL) and fetal bovine serum (10%).
2.2 Electrophysiological recordingsElectrophysiological experiments were performed as described previously.27, 28 MultiClamp-700B amplifier and Digidata-1550B A/D converter (Axon Instruments, CA, USA) were used for whole-cell patch clamp recordings. The isolated DRG neurons were transferred to a 35-mm culture dish and kept in normal external solution for at least 60 min before electrophysiological recordings. The external solution contained the following (in mM): 150 NaCl, 5 KCl, 2 MgCl2, 2.5 CaCl2, 10 HEPES, and 10 d-glucose. Its pH and osmolarity was adjusted to 7.4 with NaOH and 330 mOsm/L with sucrose, separately. Recording pipettes were pulled using a Sutter P-97 puller (Sutter Instruments, CA, USA), and its resistance was in the range of 3–6MΩ. The micropipette solution contained (in mM): 140 KCl, 2 MgCl2, 11 EGTA, 10 HEPES, 4 ATP, and 0.3 Na2GTP. Its pH and osmolarity was adjusted to 7.2 with KOH and 310 mOsm/L with sucrose, separately. After whole-cell configuration established, 70–80% series resistance and membrane capacitance current were compensated. The recording currents were sampled at 10 kHz and filtered at 2 kHz. DRG neurons (15–35μm in diameter) are used for electrophysiological recording. The membrane potential of neurons was clamped at −60 mV. Only DRG neuron with a resting membrane potential less than −50 mV was used for current-clamp recordings.
2.3 Drug applicationAll drugs were obtained from Sigma (St. Louis, MO, USA). The working concentration of drugs was freshly prepared in normal external solution. Each working drug was stored in a series of independent reservoirs and applied by gravity. The distance was ~30 μm between drug exit and recorded neurons. To block G protein and intracellular signal, some antagonists or blockers were dissolved in the internal solution and applied for intracellular dialysis through recording patch pipettes as described previously.28, 29 To ensure that dialysis drugs are infused into the cell interior, current recording was performed at least 30 minutes after cell membrane rupture.
2.4 Nociceptive behavior induced by acetic acid in ratsMale rats were first habituated for 30 minutes in a Plexiglas chamber during the nociceptive behavior experiment. After coding, rats in different groups were intraplantarly pre-treated with vehicle, different dose (10, 30, and 100 ng in 50 μl) of DEX, or 150 ng BRL44408 + 100 ng DEX, separately. After 10 min, another experimenter injected α,β-methylene-ATP (α,β-meATP, 50 μg in 50 μl) into the ipsilateral hindpaw. In one group, α,β-meATP was injected into one hindpaw and DEX (100 ng) was injected into contralateral hindpaw, and nociceptive behaviors (ie, number of flinches) were monitored over a 10-min period starting immediately after the injection of α,β-meATP. Meanwhile, mechanical allodynia was measured by paw withdrawal threshold (PWT).30 PWT of the ipsilateral hind plantar uses a series of von Frey filaments (Stoelting, Wood Dale, IL) at 0.5, 2.5, 5, and 24h after α,β-meATP injection.
2.5 Data analysisAll data were expressed as mean ± SEM. The normality of the data distribution was analyzed by the Shapiro-Wilk test. If data were normally distributed, the data were statistically compared using Student's t test or analysis of variance (ANOVA), followed by Bonferroni's post hoc test. Nonlinear curve-fitting program ALLFIT was used for statistical analysis of concentration-response data.
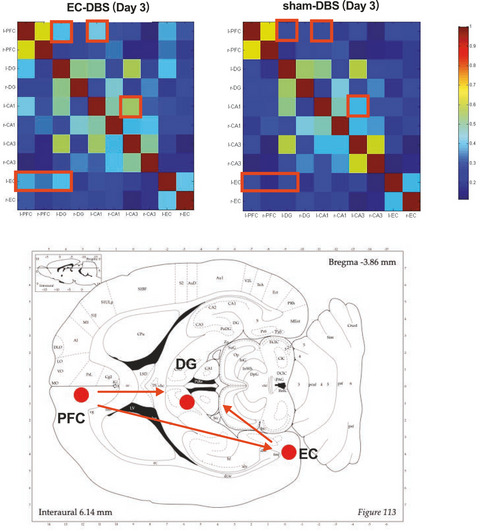
3 RESULTS 3.1 DEX inhibits P2X3 receptor-mediated ATP currents in rat DRG neurons in a concentration-dependent mannerIn the majority of small- and medium-sized (15–40 μm in diameter) DRG cells (66.7%, 6/9), an application of α,β-methylene-ATP (α,β-meATP, 100 μM) or ATP (100 μM) evoked a rapid inward currents (IATP) at a holding potential of −60 mV (Figure 1A). These α,β-meATP- and ATP-activated currents were blocked by the specifical P2X3 or P2X2/3 receptor antagonist A-317491(300 μM), but not by the P2X4 receptor antagonist PSB-12062 (10 μM) and the potent P2X7 receptor antagonist A438079 (1 μM).21 Considering that α,β-meATP is only an activator of P2X3 and P2X1 receptors, we thus concluded that the α,β-meATP-activated currents were P2X3 or P2X2/3 receptor-mediated ATP currents.31 The α,β-meATP-evoked inward currents were characterized by rapid desensitization with a mean inactivation time constant of 2068 ± 361 ms (n = 8).

DEX-induced inhibition of P2X3 receptor-mediated ATP currents in DRG neurons. (A) Two original currents were evoked by 100 μM α,β-meATP and 100 μM ATP, separately, in the same DRG cell. These currents were blocked by 300 μM A-317491 (a specific P2X3 and P2X2/3 receptor antagonist), but not by 10 μM PSB-12062 (a P2X4 receptor antagonist) and 1 μM A438079 (a potent P2X7 receptor antagonist). Membrane potentials were clamped at −60 mV. (B) Pre-application of DEX (3 μM for 5 min) to a DRG cell inhibited 100 μM α,β-meATP- and 100 μM ATP-induced currents similarly. (C) The sequential current traces illustrated that the amplitude of the 100 μM α,β-meATP-induced currents progressively decreased after a representative DRG cell was pre-treated with increasing concentrations of DEX. DEX was pre-applied to DRG tested for 5min. (D) The graph showed the concentration-effect curve of DEX on inhibition of 100 μM α,β-meATP-induced currents (IATP). The IC50 value of the curve was 1.12 ± 0.16 μM. Each point represents the mean ±SEM of 8–10 cells
In some DRG neurons, we pre-treated with DEX for 5min prior to the next IATP recording. As shown in Figure 1B, DEX pretreatment (3 μM) decreased the peak amplitudes of both α,β-meATP- and ATP-activated currents. Figure 1C shows that the peak amplitudes of 100 μM α,β-meATP-activated currents decreased as the concentration of DEX increased from 0.1 μM to 10 μM in a representative DRG cell. Figure 1D shows concentration-effect curve of DEX on IATP with an IC50 (half-maximal effective concentration) value of 1.12 ± 0.16 μM. The results suggested that DEX inhibited P2X3 receptor-mediated ATP currents in a concentration-dependent manner.
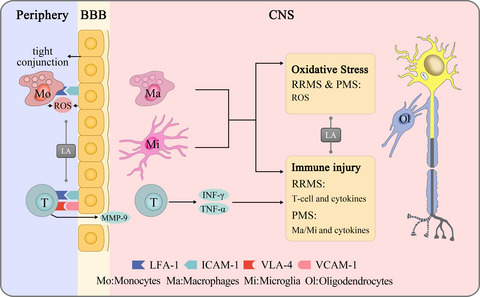
3.2 Concentration-response and current-voltage relationships for α,β-meATP in the absence and presence of DEXWe then studied whether the DEX-induced inhibition depended on the concentration of α,β-meATP. Concentration-response curves were plotted through a series of different concentration of α,β-meATP. Figure 2A shows that three representative ATP currents evoked by α,β-meATP at 3, 30, and 300 μM were decreased after DEX pretreatment (3 μM) for 5 min. Figure 2B shows that concentration-response curves for α,β-meATP were fit with the Hill equation in the absence and presence of DEX (3 μM). We observed that DEX pretreatment shifted downwards the concentration-response curve for α,β-meATP. First, maximal current response, which was evoked by 300 μM α,β-meATP, decreased 50.43 ± 4.75% with DEX (3 μM) pretreatment. Second, the Hill coefficient or slope of curves without and with DEX pretreatment was 1.40 ± 0.26 and 1.32 ± 0.37, respectively, with no significant difference (p > 0.1, Bonferroni's post hoc test). Third, DEX pretreatment did not change the EC50 of α,β-meATP for P2X3 receptors, which were 29.45 ± 1.26 μM and 31.03 ± 1.49 μM, respectively, in the absence and presence of DEX (p > 0.1, Bonferroni's post hoc test). These results suggested that DEX could inhibit the maximum response to α,β-meATP, but not shift the sensitivity of P2X3 receptors to α,β-meATP.

Concentration-response and current-voltage (I–V) relationships for α,β-meATP in the absence and presence of DEX. (A) Sequential currents were evoked by three different concentrations of α,β-meATP before and after the pre-application of DEX (3 μM for 5 min). (B) The concentration-response curves for α,β-meATP were shifted downwards in the presence of DEX (3 μM). Each point represents the mean ±SEM. of 7–10 cells. All current values from the same cell were normalized to the current response, which was induced by 300 μM α,β-meATP applied alone in the absence of DEX (marked with asterisk). (C) Sequential currents were evoked by 100 μM α,β-meATP at three different clamped potentials before and after the pre-application of DEX (3 μM for 5 min). (D) The I-V curves for 100 μM α,β-meATP-induced currents (IATP) in the absence and presence of DEX (3 μM). All current values from the same cell were normalized to the current response induced by 100 μM α,β-meATP applied alone at the holding potential of −60 mV (marked with asterisk). Each point represents the mean ±SEM of 7–10 cells. The experiment was carried out using recording pipettes filled with CsCl containing internal solution
To investigate whether the inhibition of ATP currents by DEX depended on membrane potentials, we observed the inhibitory effect of DEX on α,β-meATP-activated currents recorded at different clamping potentials. Figure 2C shows that DEX pretreatment (3 μM for 5min) inhibited the peak amplitudes of the three IATP, which were evoked by 100 μM α,β-meATP when the membrane potential was clamped at −80 mV, −40 mV, and +20 mV, separately. Figure 2D shows the current-voltage (I–V) curves for α,β-meATP with and without DEX pretreatment. DEX did not change the reversal potential (near 0 mV) of the I-V curve, but decreased the slope of curve. There was no significant difference in the DEX-induced inhibition of ATP currents at different clamping potentials from −80 to 20 mV (p > 0.1, Bonferroni's post hoc test). The results indicated that DEX voltage independently inhibited P2X3 receptor-mediated ATP currents.
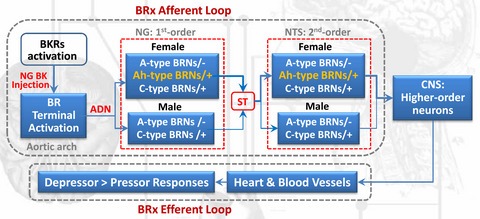
3.3 DEX inhibits ATP currents via an α2-ARs, Gi/o proteins and cAMP signaling pathwayAs a selective α2-AR agonist, is the inhibitory effect of DEX on ATP currents mediated by α2-ARs? We examined the effects of the α2-AR antagonist yohimbine and the α2A-AR antagonist BRL44408 on suppression of ATP currents by DEX. As shown in Figure 3A and B, unlike a decrease of 51.32 ± 4.63% in the IATP amplitude after 3 μM DEX pretreatment alone, the IATP amplitude decreased only 3.33 ± 1.24% and 3.72 ± 1.93 when 3 μM DEX was co-treated with 3 μM yohimbine or 3 μM BRL44408. (p < 0.01, compared with DEX pretreatment alone, one-way ANOVA followed by post hoc Bonferroni's test, n = 10). In other words, yohimbine and BRL44408 significantly blocked the inhibition of DEX on IATP. The results indicated that DEX inhibited ATP currents through α2A-ARs in DRG neurons.

Participation of α2A-ARs, Gi proteins, and cAMP signaling in the DEX-induced inhibition of ATP currents. Representative current traces in (A) and the bar graph in (B) showed IATP were inhibited by DEX (3 μM) pre-applied alone for 5 min in DRG cells, and the DEX-induced suppression of IATP was blocked by the co-application of the α2-AR antagonist yohimbine (3 μM) or the α2A-AR antagonist BRL44408 (3 μM). Currents in (B) were normalized to the control (100%). **p < 0.01, Bonferroni's post hoc test, compared with DEX column. n = 10 in each column. The current traces in (C) and the bar graph in (D) showed DEX (3 μM) had little effect on IATP in recording pipettes filled with PTX (1 μg/ml), forskolin (0.1 μM), or 8-Br-cAMP (1 mM) containing internal solution conditions, which was different from the inhibitory effect under normal internal solution conditions. However, intracellular application of BAPTA (10mM, a chelator of calcium ions) was unable to reverse the inhibitory effect of DEX on IATP. **p < 0.01, Bonferroni's post hoc test, compared with normal column. n = 10 in each column
α2A-AR is coupled with Gi/o member of the G protein family, through which it can inhibit adenylyl cyclase and attenuate intracellular cAMP levels.32, 33 First, we explored whether Gi/o proteins contribute to the DEX-induced suppression of ATP currents. A Gi/o protein inhibitor, pertussis toxin (PTX, 1 μg/mL), was applied internally to DRG neurons before DEX treatment. Figure 3C and D shows that 3 μM DEX failed to inhibit IATP in PTX-treated cells, suggesting the DEX-induced inhibition was significantly prevented by PTX. Second, we further explore intracellular signal transduction mechanisms underlying the DEX-induced suppression. The adenylate cyclase activator forskolin or the cAMP analog 8-Br-cAMP was applied internally to DRG cells through recording patch pipettes. Unlike a significant decrease in IATP amplitude under the normal internal solution conditions, 3 μM DEX produced only decreases of 2.65 ± 5.58% and 5.21 ± 8.03% on IATP, separately, after forskolin (0.1 μM) or 8-Br-cAMP (1 mM) was applied internally to DRG cells (p < 0.01, compared with normal internal solution, one-way ANOVA followed by post hoc Bonferroni's test, n = 10; Figure 3C and D). In contrast, intracellular application of BAPTA (10 mM, a chelator of calcium ions) was unable to reverse the inhibitory effect of DEX on IATP. These results suggested that DEX inhibited ATP currents via a Gi/o proteins and intracellular cAMP signaling pathway.
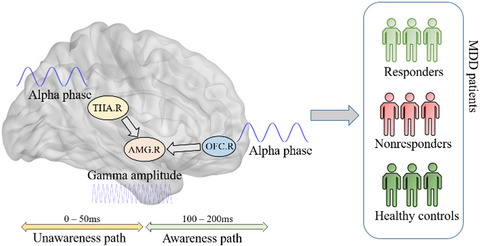
3.4 DEX suppresses α,β-meATP-evoked action potentials in rat DRG neuronsWe further investigated the effect of DEX on action potentials (APs) triggered by α,β-meATP. In the same DRG cell, 100 μM α,β-meATP not only induced a large inward current under voltage-clamp conditions but also evoked burst of APs under current-clamp conditions (Figure 4A and C). Consistent with the results observed under voltage-clamp conditions, DEX also exerted an inhibitory effect on α,β-meATP-evoked APs. As shown in Figure 4B, DEX (3 μM for 5 min) pretreatment significantly decreased the number of APs triggered by α,β-meATP in six DRG cells (p < 0.01, paired t test). However, co-application of 3 μM BRL44408 and 3 μM DEX failed to decrease the number of APs in other six DRG cells (p ˃ 0.1, paired t test; Figure 4D). The results suggested that DEX also suppressed α,β-meATP-evoked APs through α2A-ARs in rat DRG neurons.

DEX-induced suppression of α,β-meATP-evoked action potentials in rat DRG neurons. Original traces in (A) and (C) showed application of 100 μM α,β-meATP to the same DRG cell caused an inward current and action potentials (APs) under voltage-clamp and current-clamp conditions, respectively. Original APs were recorded before and after application of DEX (3 μM for 5 min) alone (A) or co-application of both DEX (3 μM) and BRL44408 (BRL, 3 μM) (C) in two different DRG neurons. Data in (B) and (D) showed application of DEX alone, but not co-application of both DEX and BRL44408, significantly decreased the number of APs evoked by 100 μM α,β-meATP. ** p < 0.01, paired t test, n = 6 cells
3.5 DEX relieves α,β-meATP-induced nociceptive behaviors in ratsFinally, we investigated whether the suppression of P2X3 receptors by DEX in vitro played a role in the nociceptive behaviors induced by α,β-meATP in vivo. Figure 5A shows intraplantar injection of α,β-meATP (50 μg in 50 μl) caused an intense spontaneous flinch/shaking response in rats, which was attenuated by intraplantar pretreatment of DEX. DEX dose dependently (10, 30, and 100 ng) relieved the nociceptive behaviors induced by α,β-meATP (p < 0.05 and 0.01, one-way ANOVA followed by post hoc Bonferroni's test, n = 10; Figure 5A). The anti-nociceptive effect of 100 ng DEX was blocked by co-treated 150 ng BRL44408 (p < 0.01, one-way ANOVA followed by post hoc Bonferroni's test, n = 10; Figure 5A). In addition, 100 ng DEX had no effect on the α,β-meATP-induced nociceptive behaviors when injected into the contralateral paws. The results suggested that DEX had an anti-nociceptive effect on the α,β-meATP-induced nociceptive behaviors in vivo through peripheral α2A-ARs.

Relief of α,β-meATP-evoked nociceptive behaviors by DEX in rats. (A) Intraplantar injection of α,β-meATP (50μg in 50μl) caused spontaneous flinching behaviors in rats. Intraplantar pretreatment of DEX (10, 30, and 100 ng) dose dependently decreased the number of α,β-meATP-induced flinching behaviors. The anti-nociceptive effect of DEX (100ng) on the flinching behaviors was completely prevented by co-treatment of the α2A-AR antagonist BRL44408 (BRL, 150 ng). DEX (100 ng) had no effect on α,β-meATP-induced flinching behaviors when it was injected into the contralateral hindpaw and α,β-meATP was injected into one hindpaw (100 ng contral). Bonferroni's post hoc test, *p < 0.05, **p < 0.01, compared with vehicle column; ## p < 0.01, compared with 100 ng DEX column. Each column represents the mean ±S.E.M. of 10 rats. (B) Intraplantar injection of α,β-meATP (50 μg in 50 μl) also caused a remarkable decrease in paw withdrawal thresholds (PWT, in g) at 0.5 and 2.5 h after injection and recovery at 24 h. The α,β-meATP-induced mechanical allodynia was significantly relieved by intraplantar pretreatment of DEX (100 ng), but not co-treatment of DEX (100 ng) and BRL44408 (BRL, 150 ng). *p < 0.05, **p < 0.01, Bonferroni's post hoc test, n = 10 rats in each group
We also observe that the effect of DEX on the mechanical allodynia induced by α,β-meATP in rats. Figure 5B shows intraplantar injection of α,β-meATP (50 μg in 50 μl) resulted in a significant decrease in the paw withdrawal threshold (PWT) within 0.5 and 4 h after injection, and recovery at 24 h. Intraplantar pretreatment of DEX had also an analgesic effect on the mechanical allodynia. The α,β-meATP-induced mechanical allodynia significantly decreased within 0.5 and 4 h after 100 ng DEX pretreatment (p < 0.05 and 0.01, Bonferroni's post hoc test, compared with vehicle +α,β-meATP group, n = 10 rats; Figure 5B). The analgesic effect of DEX was completely blocked by co-treated 150 ng BRL44408 (p < 0.05 and 0.01, Bonferroni's post hoc test, compared with DEX +α,β-meATP group, n = 10 rats; Figure 5B). The results suggested that DEX had also an analgesic effect on the α,β-meATP-induced mechanical allodynia through peripheral α2A-ARs.
4 DISCUSSIONThe present data demonstrated that the selective α2-AR agonist DEX suppressed the electrophysiological activity of P2X3 receptors in rat DRG neurons. DEX reduced not only the amplitude of ATP currents but also the action potential bursts induced by α,β-meATP. The α2A-ARs, PTX-sensitive Gi/o proteins, and cAMP signaling cascades were involved in the inhibition of P2X3 receptors by DEX. Behaviorally, DEX also relieved P2X3 receptor-mediated nociceptive behaviors in rats by activating peripheral α2A-ARs.
The recorded ATP currents in the present experiments were mediated by P2X3 receptors, because they could be blocked by specifical antagonist of P2X3 and P2X2/3 receptor A-317491.21 Moreover, α,β-meATP can only activate P2X3 and P2X1 receptors.31 ATP receptors include P2X1-7 subtypes. Among all subtypes, P2X3 receptor subtype is mainly located in a subset of small- and medium-sized nociceptive DRG neurons.17-19 The present study showed that DEX, a selective α2A-AR agonist, concentration dependently inhibited P2X3 receptor-mediated ATP currents. The DEX-induced suppression did not alter the sensitivity of P2X3 receptor to α,β-meATP, but decreased the maximum response to α,β-meATP. DEX suppressed ATP currents in voltage-independent manner. P2X3 receptor is a cation-permeable channel. The activation of P2X3 receptors by α,β-meATP evokes a rapid inward current sufficient to induce membrane depolarization and to generate firing.31, 34 Under the current-clamp conditions, DEX also suppressed the number of APs evoked by α,β-meATP in rat DRG neurons. Obviously, the two results corroborated each other in the current-clamp and voltage-clamp experiments.
DEX suppressed ATP currents through α2A-ARs, since DEX-induced inhibition was mimicked by another α2-AR agonist clonidine and completely blocked by the α2A-AR antagonist BRL44408. All three subtypes of α2A-, α2B-, and α2C-AR have been identified in DRGs.4 Subtypes of α2A- and α2C-AR are expressed in most DRG neurons, while is found only in a very small population.3, 4, 35 In addition, both DEX and clonidine have more specific activity against α2A-AR subtype.36 Considering that the α2A-ARs maybe co-express with P2X3 receptors in DRG neurons, it was completely possible that DEX decreased ATP currents through α2A-ARs. The present study showed that the inhibition of ATP currents by DEX / clonidine only occurred in some but not all DRG neurons, which may be related to the degree of the co-existence of α2A-ARs and P2X3 receptors in rat DRG neurons, although the evidence of morphological co-existence remains to be elucidated. It has been shown that the activation of α2A-AR subtypes also modulates the functional activity of other cation channels, such as Nav1.8, TRPV1, TRPM8, and ASICs in DRG neurons.14, 16, 29, 37
α2A-AR belongs to Gi/o protein-coupled receptor family that can inhibit adenylyl cyclase and attenuate intracellular cAMP levels.32, 33 The present data showed that Gi/o proteins and intracellular cAMP signaling were involved in the DEX-induced suppression of ATP currents, as demonstrated by the results that the DEX-induced inhibition was lack after DRG neurons were intracellularly dialyzed with the Gi/o protein inhibitor PTX, the adenylate cyclase activator forskolin, or the cAMP analog 8-Br-cAMP. Previous studies have showed that P2X3 receptor-mediated ATP currents are regulated by intracellular cAMP/PKA signaling. Ma group reported that 17β-estradiol and progesterone rapidly attenuate ATP currents in DRG neurons via a cAMP-PKA signaling pathway.38, 39 On contrary, PGE2 enhances P2X3 receptor-mediated currents in DRG neurons via its EP3 receptor and a cAMP/PKA-dependent signaling pathway.40 It has been also reported that PKC and PKA potentiate α,β-meATP-induced currents in rat DRG neurons in a synergistic manner.41 Obviously, the latter two reported studies are consistent with the current results that the activation of α2A-ARs by DEX reduced ATP currents through down-regulating intracellular cAMP signaling pathway. Similarly, cannabinoid inhibits ATP-induced currents and calcium influx through Gi/o protein-coupled CB1 cannabinoid receptors and AC-cAMP-PKA signaling pathway in rat trigeminal ganglionic neurons and cultured spinal dorsal horn neurons.42, 43 In addition, intracellular application of BAPTA, a chelator of calcium ions, failed to reverse the inhibitory effect of DEX on α,β-meATP-induced currents, suggesting no involvement of intracellular Ca.2+
P2X3 receptors are not only expressed in the somata of DRG neurons but also expressed in peripheral nociceptive sensory nerve endings. Injection of ATP into the skin elicits pain via P2X3 receptors.44 Intraplantar injection of P2X3 receptor agonists results in spontaneous pain behaviors and mechanical allodynia in rats.25, 45, 46 Local pretreatment of α1-AR agonists augment P2X3 receptor-mediated flinching behaviors in rats.47 This behavioral finding is consistent with electrophysiological results that the activation of α1-ARs by noradrenaline potentiates ATP-evoked currents in DRG neurons by activating PKC.26 The present results showed that peripheral pretreatment of the DEX dose dependently relieved the α,β-meATP-induced nociceptive behaviors. The anti-nociceptive effect of DEX occurred locally rather than systematically by directly activating peripheral α2A-ARs localized on nociceptors. First, treatment of contralateral hindpaws with DEX failed to relieve the α,β-meATP-induced nociceptive behaviors. Second, the anti-nociceptive effect of DEX was abolished by local treatment with BRL44408, an α2A-AR antagonist. In further mechanical allodynia experiments, DEX had also an analgesic effect on the α,β-meATP-induced mechanical allodynia through peripheral α2A-ARs. The present behavioral results apparently confirmed the aforementioned electrophysiological data and vice versa. Somata of DRG neurons in the electrophysiological experiments was used as a model to observe the characteristics of peripheral terminals. It may occur that the activation of α2A-ARs by DEX suppressed co-existed P2X3 receptors in peripheral nociceptive sensory nerve endings, which could reduce P2X3 receptor-mediated currents and action potential bursts, and then result in attenuated nociceptive behaviors.
5 CONCLUSIONSUnder inflammatory and neuropathic pain conditions, not only a large amount of ATP is released from damaged cells but also total expression and membrane expression of P2X3 receptors increases in DRG neurons.25, 48-50 These P2X3 receptors, activated by released ATP, plays a prominent role in some pain states.51 Our results suggested that DEX suppressed P2X3 receptor-mediated the electrophysiological activity and nociception through α2A-ARs, revealing a peripheral novel mechanism underlying the analgesia of DEX. Clinically, DEX, even when applied locally, may also effectively relieve pain involving peripheral P2X3 receptors.
ACKNOWLEDGMENTThis work was supported by the National Natural Science Foundation of China (No. 81671101) and Science and Technology Research Project of Hubei Provincial Department of Education (No. Q20212802).
CONFLICT OF INTERESTAll authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONSWPH designed this research. JWH, WLQ, QL, SW, TTL, and CYQ performed the experiments. JWH, WLQ, and QL participated in data analysis. JWH, WLQ, QL, and WPH wrote the paper. All authors contributed substantially to this research and reviewed this manuscript.
ETHICAL APPROVALThe animal study was reviewed and approved by the animal research ethics committee of Hubei University of Science and Technology.
Comments (0)