
Remember me









The UPBEAT study is a multi-institutional phase II study registered in the UMIN Clinical Trials Registry (UMIN000047080) on March 4, 2022. The protocol for this study was approved by the Institutional Review Board of Kobe City Medical Center General Hospital and has been reported previously [14]. An overview of this trial is shown in Fig. 1. Herein, we enrolled patients who had previously undergone BCS and treated them with ultra-HF-WBI as a protocol treatment. Upon enrolling 28 patients, patient registration was paused for three months. Following this, we conducted a preplanned interim analysis that evaluated AEs after three months of follow-up to determine the safety of proceeding with the trial.
Fig. 1
Japanese women who had previously undergone BCS were registered and received ultra-HF-WBI of 26 Gy in five fractions over one week. When the number of registered patients reached 28, patient enrollment was paused. An interim analysis was performed after three months of follow-up to determine whether it was safe to continue the study, up to a total of 98 patients. BCS breast-conserving surgery, HF hypofractionated, WBI whole-breast irradiation, AE adverse event
PatientsEligible patients were required to meet all of the following criteria: no distant metastasis on any examination before surgery; had undergone BCS for breast cancer; pathological eligibility criteria, including invasive carcinoma or ductal carcinoma in situ (DCIS), pathological stage of Tis–T3 N0–N1 (including Tis Nx), and negative margin as no tumor on ink; radiation therapy initiated within 70 days of BCS, or if additional resection was performed, within 70 days of additional resection and 140 days of BCS, or within 42 days of the last day of adjuvant chemotherapy; Asian ethnicity and female sex; age ≥ 40 years at the time of registration; Eastern Cooperative Oncology Group (ECOG) performance status of 0–1; no prior radiation therapy administered to the chest except to the contralateral breast; and written informed consent provided by the patient in Japanese. The exclusion criteria are described in the previously published study protocol [14]. In the current study, all staging was based on the eighth edition of the TNM Classification of Malignant Tumours by the Union for International Cancer Control.
TreatmentAll patients underwent computed tomography (CT) simulations in the supine position, with the ipsilateral arm or both arms elevated. A patient-fixing device was mandatorily used for this purpose. For CT simulation, radiopaque markers were used to identify the boundaries of the breast. For spatial localization during each treatment, megavoltage electronic portal imaging or a similar imaging system was used. During the simulation process and each treatment session for patients with cancers of the left breast, deep inspiration breath-holding was recommended to reduce the dose delivered to the heart [15, 16]; however, this was not essential, and free breathing was permitted in all cases.
The target volumes were delineated as follows: the clinical target volume (CTV) included the whole of the conserved breast, excluding the muscle and the underlying rib cage; the planning target volume (PTV) included the CTV plus a typical margin of 5 mm; the PTV for a dose–volume histogram (PTV_DVH), defined to evaluate the PTV dose, was cropped to 5 mm inside the skin and 5 mm from the lung surface. The major organs at risk in ultra-HF-WBI were the ipsilateral lung and heart. Dosimetric criteria were established (Online Resource 1) to ensure the quality of radiation treatment planning. The use of wedge filters (including dynamic wedges) was recommended, and the field-in-field technique was permitted. The use of 4–10 MV X-rays was mandatory, whereas the use of a bolus to the skin was not permitted. Boost irradiation and supraclavicular irradiation were also not permitted.
Ultra-HF-WBI was delivered to the whole breast after BCS at a daily dose of 5.2 Gy, once per day, five times per week, and up to a total of 26 Gy in five fractions over a recommended treatment period of ≤ 10 days. Hormonal and/or anti-HER2 therapies were permitted during the treatment period; however, chemotherapy was not allowed.
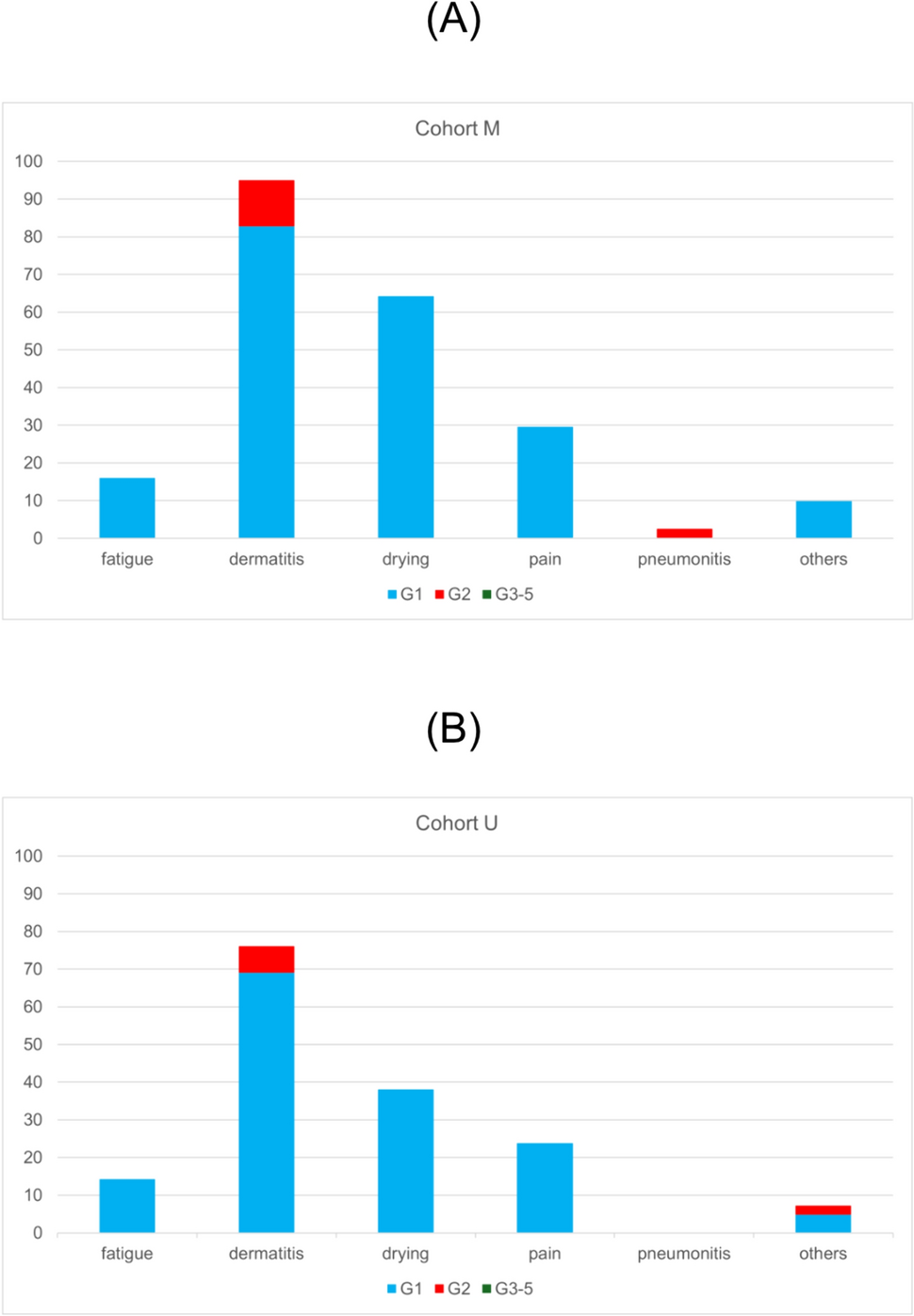
Endpoints and assessment methodsIn this interim analysis, the endpoint was the rate of acute AEs of grade ≥ 2. Acute AEs were defined as toxicities experienced within 90 days of initiating radiation therapy, and were evaluated using the Common Terminology Criteria for Adverse Events (CTCAE) v5.0. The evaluation was performed immediately after treatment (within two weeks following the completion of WBI) and three months following the initiation of WBI (with an acceptable scale of one month before and after). The primary objective of our study was to determine the most frequent AEs experienced following WBI—namely, radiation dermatitis (including erythema and desquamation), pneumonitis, pain (including chest wall pain, breast pain, and skin pain), fracture, telangiectasia, superficial soft tissue fibrosis, pericarditis, and myocardial infarction.
Statistical analysisFor the interim analysis, the required sample size was calculated to be 25 patients, with a one-sided alpha level of 10%, power of 80%, threshold value of 30%, and expected value of 12.4%, which was determined based on the proportion of acute AEs of grade ≥ 2 (38/306) reported in the JCOG0906 study [4, 5]. To account for deviations from the prescribed procedure, the total sample size in the interim analysis was estimated to be 28.
We considered that if acute AEs of grade ≥ 2 were found in ≤ 5 patients, it would represent an acceptable proportion and patient registration could be resumed. Otherwise, we would conclude that ultra-HF-WBI were markedly unsafe in Japanese women, and the study would be terminated.
Comments (0)