
記住我



Testicular sex cord–stromal tumors (TSCSTs) are relatively rare, comprising ~5% of all testicular tumors and a slightly higher proportion of testicular neoplasms in pediatric patients.1–4 Historically, pure TSCSTs included 3 major entities: Leydig cell tumor, Sertoli cell tumor, and granulosa cell tumor.5 Lesions that did not fit into these diagnostic categories were often considered “unclassified” TSCSTs. In recent years, immunophenotypic and molecular analyses of TSCSTs have led to a more detailed understanding of their biological and genomic features, resulting in the incorporation of additional entities to the latest WHO classification (Table 1).6
TABLE 1 - Sex Cord–stromal Tumor Types Included in the Fifth Edition of the World Health Organization Classification of Urinary and Male Genital Tumors Leydig cell tumorNOS indicates not otherwise specified.
Notwithstanding their rarity, TSCSTs are clinically relevant for several reasons. First, ~90% of TSCSTs across different histologic subtypes are clinically indolent and can be safely managed with testis-sparing surgery.7 Therefore, their proper identification in intraoperative biopsies is important to avoid overtreatment. Second, some TSCSTs are associated with disorders of sex development and inherited cancer syndromes.8–11 Hence, recognition of these subtypes may lead to the identification of an underlying hereditary condition with a significant impact on patients and their families. Third, malignant TSCSTs are typically resistant to systemic treatment but may benefit from timely surgical intervention.7,12 Hence, identifying TSCST with malignant potential in radical orchiectomies is of paramount importance to determine which patients may require upfront retroperitoneal lymph node dissection. Each point mentioned above is associated with problems that are intrinsic to the clinical and biological characteristics of TSCSTs. From a practical standpoint, most pathologists have limited experience with these rare tumors; therefore, cases without typical morphologic features often require workup with immunohistochemistry and cannot be recognized intraoperatively. Moreover, TSCST types associated with inherited cancer syndromes may require assessment with immunohistochemistry and molecular techniques that are not readily available in all practice settings.13–15 Finally, perhaps the most important clinical problem is that the biological potential of a subset of TSCSTs may be difficult to predict based on the histopathologic features of the primary tumor.11
In recent years, several studies have assessed the molecular and clinical characteristics of TSCSTs in an attempt to overcome some of the aforementioned problems. This has led to the identification of biomarkers that could be useful for prognostication and diagnosis. Moreover, molecular analyses have identified potentially new or refined TSCST types, while questioning the validity of some entities currently included in the WHO classification.16–18 This review summarizes novel histopathologic, clinical, and molecular findings that may lead to a reappraisal of established concepts and improve the diagnostication and clinical management of TSCSTs in the coming years.
LEYDIG CELL TUMORLeydig cell tumor is the most common TSCST type in both adult and pediatric patients.5,19,20 It demonstrates a biphasic incidence, with peaks in early childhood and mid-adulthood.1,21,22 Malignant cases are restricted to postpubertal patients and often affect elderly men.19,22,23 Another important difference between pediatric and adult Leydig cell tumors pertains to their clinical presentation. More specifically, pediatric tumors are typically associated with isosexual precocious pseudopuberty, whereas adult patients most often present with a painless testicular mass.21,22 Tumors with overlapping morphology arising in children with congenital adrenal hyperplasia are thought to have a different cell of origin and develop in response to elevated ACTH levels, being most likely nonneoplastic in nature (currently designated “testicular tumors of the androgenital syndrome”).24
Microscopically, typical Leydig cell tumors are characterized by solid nests and sheets of polygonal neoplastic cells with an abundant amount of bright eosinophilic cytoplasm.4,21,22 Cell membranes are delicate or imperceptible, giving these tumors a somewhat syncytial appearance at low to intermediate magnification. Nuclei are round and regular, with optically clear chromatin and a single conspicuous nucleolus. “Ground-glass” chromatin and nuclear pseudo-inclusions are relatively common. Other diagnostically useful features are the presence of Reinke crystals, intracytoplasmic hyaline globules, and lipofuscin pigment. Reinke crystals, a specific marker of Leydig cell differentiation, are only seen in about a third of all Leydig cell tumors, with some studies suggesting that they are more common in nonaggressive neoplasms.19,21 Leydig cell tumors frequently exhibit a minimum amount of stroma containing delicate branching thin-walled vessels, but cases with more abundant myxoid or edematous stroma are sometimes seen. Architectural and cytomorphologic variations are not unusual, including tumor cells with lipid-laden microvesicles. The most sensitive immunomarkers for Leydig cell tumor are SF1 and inhibin alpha (positive in >95% each) and calretinin (positive in ~80%), whereas FOXL2, WT1, and SOX9 are only positive in minor subsets (0% to 20%; Table 2).25,26 Other markers frequently expressed by Leydig cell tumors include AR, Melan-A, and synaptophysin, although their clinical utility is quite limited.26 Of note, a subset expresses nuclear beta-catenin, usually with a focal or multifocal distribution (see molecular characteristics; Fig. 1).14
TABLE 2 - Relevant Immunohistochemical Features of Testicular Sex Cord–stromal Tumors Relevant positive IHC Relevant negative IHC Leydig cell tumor Inhibin (>95%), SF1 (>95%), calretinin (~80%), β-catenin (~50%, focal or multifocal), MDM2 (positive in a subset of aggressive cases) FH (loss of expression is seen in a subset of cases, associated with aggressive features in prior studies), WT1 (negative in >90%), FOXL2 (negative in >90%), SOX9 (negative in ~80%) Sertoli cell tumor, NOS WT1 (40-50%), SOX9 (~60%), FOXL2 (~50%), SF1 (~80%), β-catenin (~70%, diffuse) — Large cell calcifying Sertoli cell tumor Inhibin, calretinin, SF1, SOX9 PRKAR1A (expression is lost in >90%), β-catenin Adult granulosa cell tumor May express any of the sex cord–stromal tumor markers/no specific IHC findings — Juvenile granulosa cell tumor May express any of the sex cord–stromal tumor markers/no specific IHC findings — Myoid gonadal stromal tumor Coexpression of SMA and S100 (by definition) Calretinin, SOX9 Inflammatory and nested testicular sex–cord tumor SF1, CD30, inhibin SALL4 (important given the morphologic overlap with seminoma), β-cateninIHC indicates immunohistochemistry, NOS, not otherwise specified.
 FIGURE 1:
FIGURE 1: Beta catenin expression in Leydig cell tumors. (A, B) A subset of benign and malignant Leydig cell tumors harbor CTNNB1 variants and demonstrate nuclear staining for beta-catenin. Unlike Sertoli cell tumor, NOS, Leydig cell tumor most often exhibits focal/multifocal nuclear positivity for beta-catenin, suggestive of subclonal activation of the Wnt pathway. NOS indicates not otherwise specified.
Approximately 10% of Leydig cell tumors spread beyond the testis and follow an aggressive clinical course.19,22 In the early series, several pathologic features such as large tumor size (≥5 cm), >3 mitoses per 10 high-power fields, tumor necrosis, invasive growth, marked nuclear atypia, and lymphovascular invasion were identified as potential predictors of malignant behavior.6,22,23 In recent years, a few groups have proposed the implementation of multiparametric systems akin to those used in adrenal cortical neoplasms to predict the risk of metastases in Leydig cell tumors.19,27 Based on data from a meta-analysis, Fankhauser et al19 have found that age ≥42 years, tumor size ≥3 cm, and the presence of adverse histopathologic findings (lymphovascular invasion, necrosis, increased mitotic activity, and atypia) were associated with a higher risk of metastasis. In contrast, the presence of gynecomastia, Reinke crystals, and/or lipofuscin pigment were associated with a lower risk of metastatic spread.19 A system that incorporates patient age (≥42 y vs.<42 y), tumor size (≥3 cm vs.<3 cm), and the presence of adverse histopathologic findings as risk factors, as well as gynecomastia, the presence of lipofuscin pigment, and/or Reinke crystals as protective factors can be useful to estimate the probability of malignant behavior. In brief, the combination of 2 or more adverse clinicopathologic factors would indicate a high risk of malignant behavior, even if protective factors are concurrently present. According to this system, the presence of a single adverse clinicopathologic factor in the absence of concurrent protective factors also confers a risk of malignancy that is not insignificant, with this risk being higher for tumors ≥3 cm.19 Therefore, according to this system, the only Leydig cell tumors with a low (10% or less) risk of metastases are those that lack adverse clinicopathologic findings or, alternatively, those that exhibit only 1 adverse finding with concurrent protective factors.19 The major strengths of this system are that it integrates clinical and histopathologic variables, and it is based on data obtained from a meta-analysis comprising more than 1300 patients. Its major drawback is that it groups all adverse histopathologic findings into a single variable, artificially homogenizing their prognostic significance. For instance, based on our empirical observations, the presence of lymphovascular invasion and overt tumor necrosis is more worrisome than the presence of atypia or borderline elevated mitotic activity (3 to 4 mitoses per 10 high-power fields). Colecchia et al27 have developed a different multiparametric system that assigns 0 to 2 points for number of mitoses per 10 high-power fields (0 mitosis=0 point, 1 to 3 mitoses=1 point, and >3 mitoses=2 points), 0 to 2 points for tumor size (<1.5 cm=0 point, 1.5 to 2.5 cm=1 point, and >2.5 cm=2 points), and 1 point for the presence of each of the following findings: invasive/infiltrative growth, vascular invasion, and necrosis.27 The scale of this scoring system goes from 0 to 7, with tumor scores of 4 to 7 being associated with malignant behavior. The main limitations of this score are that it is based on a comparison of metastasizing and nonmetastasizing cases collected retrospectively, and it does not include patient age, a variable that has been associated with risk of aggressive clinical behavior in other studies. Despite their drawbacks, these multiparametric systems may help improve risk stratification of patients with Leydig cell tumors, and merit validation and further refinement in future studies. Moreover, these systems may also be useful to evaluate risk in patients with other types of TSCSTs with slight modifications. An important conclusion from both of the aforementioned studies is that the size threshold of 5 cm which had been traditionally considered a risk factor for malignant behavior is likely inappropriate, with 2.5 to 3 cm being a better cutoff to identify potentially aggressive cases.19,27
The purported driver of pediatric Leydig cell tumors was identified before the era of massively parallel sequencing. These neoplasms typically harbor a somatic gain-of-function variant in the luteinizing hormone/human choriogonadotropin receptor gene (LHCGR p.D578H; Fig. 2).28 This mutation leads to the activation of Gs signaling and subsequent proliferation of Leydig cells. A Leydig cell tumor in an adult patient with a gain-of-function GNAS mutation has also been reported, suggesting that variants in genes that code for proteins located downstream of the luteinizing hormone receptor may also be oncogenic in Leydig cell tumors.29 However, most adult Leydig cell tumors do not harbor LHCGR or GNAS mutations.15 Instead, gain-of-function CTNNB1 mutations and loss-of-function FH variants have been identified in subsets of Leydig cell tumors affecting adult patients.14,15,27,30 The CTNNB1 mutations present in Leydig cell tumors affect a region of the N-terminal domain that is critical for marking the protein for degradation and are therefore expected to have an activating effect.14 Based on the assessment of variant allele frequencies and distribution of nuclear beta-catenin staining, CTNNB1 mutations appear to be subclonal in many cases.14 Hence, they may represent a relatively late event associated with tumor progression rather than the main driver in some Leydig cell tumors. More recently, MDM2 amplification, FH inactivation, TERT fusions, and chromosomal imbalances (ie, aneuploidy) have been associated with neoplasms that metastasize and/or exhibit worrisome histopathologic features.14,27,31 Some of these alterations, such as FH deficiency and MDM2 amplification, can be assessed in routine clinical practice using immunohistochemistry or fluorescence in situ hybridization (Fig. 3), representing potential biomarkers that could be incorporated in multiparametric risk assessment systems.27
 FIGURE 2:
FIGURE 2: Molecular alterations in Leydig cell tumors. Figure created with BioRender.com.
 FIGURE 3:
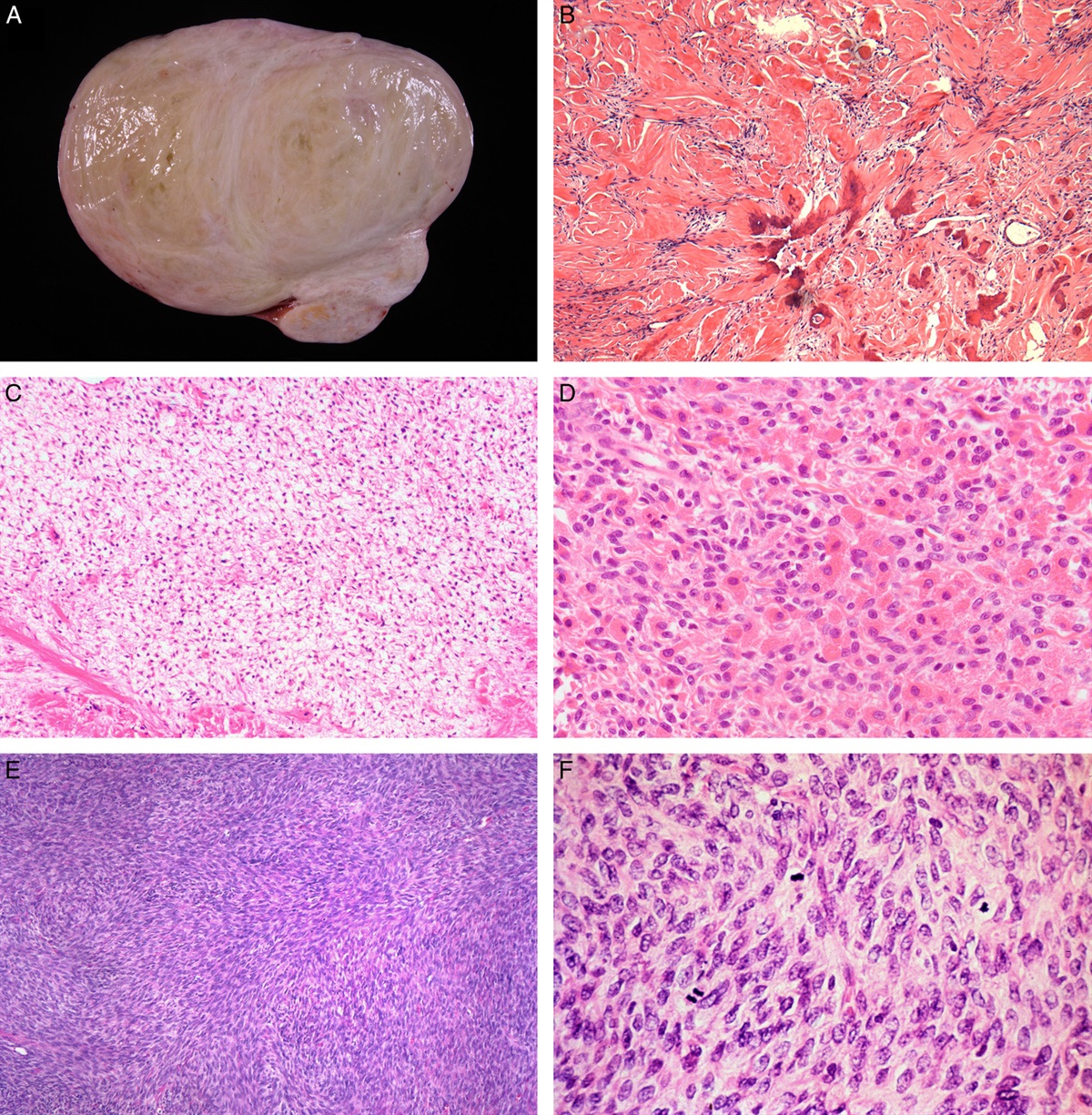
FIGURE 3: MDM2 as a marker of aggressive behavior in Leydig cell tumors. (A) Leydig cell tumor with vascular invasion. (B) This tumor is diffusely positive for MDM2 by immunohistochemistry. (C) Fluorescence insight to hybridization demonstrates the amplification of MDM2 (green probe) in another example of malignant Leydig cell tumor.
SERTOLI CELL TUMORSertoli cell tumor is the second most common type of TSCST and represents ~1% of all testicular neoplasms.1,21 It affects both prepubertal and adult postpubertal patients, and ~10% exhibit malignant clinical behavior.32 Most cases present as a painless testicular mass, with findings secondary to sex hormone production seen in a relatively minor subset (~10% to 20%).32 Although malignant cases affect mostly adult men, rare metastasizing cases have been reported in prepubertal patients.33,34 Sertoli cell tumor is a nosologic category that comprises 2 main subtypes: Sertoli cell tumor, not otherwise specified (NOS) and large cell calcifying Sertoli cell tumor (LCCSCT). These tumors have markedly different histopathologic and molecular features and will be discussed separately. Intratubular large cell hyalinizing Sertoli cell neoplasia, an exceedingly rare neoplasm occurring in patients with Peutz-Jeghers syndrome, is outside the scope of this article and the reader is referred to the original report by Ulbright et al.10
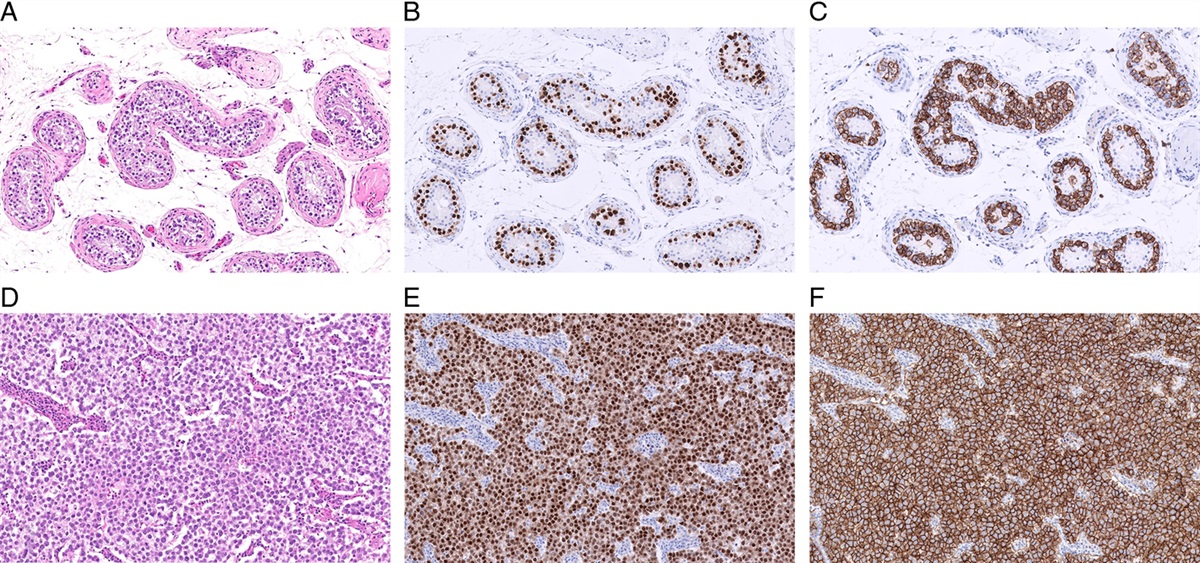
Sertoli Cell Tumor, Not Otherwise SpecifiedTypical examples of Sertoli cell tumor, NOS exhibit tubular architecture, diffusely or focally. However, these tumors often comprise multiple growth patterns, including cords, trabeculae, solid nests, and solid sheets.21,35,36 Tumor cells are epithelioid, with round to ovoid nuclei, inconspicuous nucleoli, and a moderate amount of cytoplasm that is eosinophilic, amphophilic, or clear/vacuolated. Compared to the Leydig cell tumor, Sertoli cell tumor, NOS has more abundant stroma, which is typically collagenous and may exhibit different degrees of hyalinization.36 Otherwise, typical Sertoli cell tumor, NOS may include cells with signet ring morphology, and it is currently debated whether tumors with pure or extensive signet ring cell features should be considered Sertoli cell tumor, NOS, or a different tumor type.37–40 In the latest WHO classification of genitourinary tumors (2022), TSCSTs with pure signet ring cell morphology are considered a distinct entity (signet ring stromal tumors). Malignant cases typically exhibit worrisome histopathologic features, including nuclear atypia/pleomorphism, invasion of extratesticular structures, elevated mitotic counts, large size, and necrosis.6,21,35 It has been well recognized that the morphologic features of a subset of malignant Sertoli cell tumor, NOS overlap with those of seminoma.41 More specifically, these neoplasms show a solid growth pattern, lesional cells with vacuolated (clear) cytoplasm, and a prominent lymphoplasmacytic infiltrate.41 While historically considered examples of malignant Sertoli cell tumor, new data suggest that they may represent a distinct entity (see discussion about their molecular characteristics below).16 Sertoli cell tumors, NOS are often positive for SF1 (~80%) and SOX9 (~60%), whereas the expression of WT1, calretinin, inhibin alpha, and FOXL2 is variable (~40% to 50% each).36 Beta-catenin stains ~70% of cases overall (Fig. 4), being more sensitive for benign cases (~90%).25,42–44 Other markers, such as synaptophysin, AR, and keratins, may also be positive but are not clinically useful.
 FIGURE 4:
FIGURE 4: Beta catenin expression in Sertoli cell tumor, NOS. (A, B) Diffuse nuclear expression of beta-catenin is seen in ~70% of Sertoli cell tumor, NOS overall (>90% in benign cases). NOS indicates not otherwise specified.
Approximately 70% of all Sertoli cell tumor, NOS harbor gain-of-function CTNNB1 variants.42 These variants are clustered within a specific region of exon 3, which codes for a critical region of the N-terminal domain of the protein.14,42,45 Specifically, this region of the N-terminal domain contains Serine and Threonine residues that are phosphorylated by CK1 and GSK3β and mark the protein for proteasomal degradation. Gain-of-function exon 3 CTNNB1 mutations typically alter these critical phosphorylation sites, making the protein resistant to degradation and available for activation of Wnt signaling.45 Alternatively, some Sertoli cell tumors, NOS harbor loss-of-function APC variants that also result in decreased degradation of beta-catenin and upregulation of Wnt signaling.44CTNNB1 or APC mutations are found in more than 90% of primary tumors and in ~50% of metastasizing tumors, which correlates well with the lower rate of nuclear beta-catenin expression seen in malignant Sertoli cell tumor, NOS.42–44 Of note, malignant Sertoli cell tumors with CTNNB1 mutations also exhibit additional genomic alterations such as multiple chromosomal imbalances (ie, aneuploidy), which are likely associated with biological progression (Fig. 5).30,44 Until recently, only 2 cases of Sertoli cell tumor, NOS arising in patients with familial adenomatous polyposis (FAP) had been documented, but the association was largely anecdotal, because there was no evidence that a germline APC variant was implicated in the oncogenesis of these neoplasms.46,47 This year, Siegmund and colleagues analyzed 3 Sertoli cell tumors, NOS (including 2 occurring in patients with a clinical history of FAP) and 1 malignant unclassified TSCST with APC variants, demonstrating that germline loss-of-function APC variants with loss-of-heterozygosity were the underlying oncogenic mechanism in these tumors48. This suggests that FAP and variants thereof may predispose patients to develop TSCSTs in general, and Sertoli cell tumor, NOS in particular. Sertoli cell tumors with APC mutations are morphologically identical to those with CTNNB1 mutations and express nuclear beta-catenin, hindering the identification of cases potentially associated with germline APC variants. Until further data are generated, the presence of bilateral or multifocal testicular lesions and a history of other neoplasms commonly associated with FAP (eg, desmoid fibromatosis) should raise the possibility of a germline APC variant in patients with Sertoli cell tumor, NOS. Interestingly, the genomic methylation profiles of TSCSTs without worrisome histopathologic features that exhibit unusual growth patterns or mixed Sertoli-Leydig cell components and express nuclear beta-catenin diffusely are similar to those of typical Sertoli cell tumors.18 This suggests that the biologic spectrum of Sertoli cell tumor, NOS may encompass tumors with unusual growth patterns (eg, the absence of unequivocal tubular or corded components) that are driven by gain-of-function CTNNB1 variants and/or loss-of-function APC variants.
 FIGURE 5:
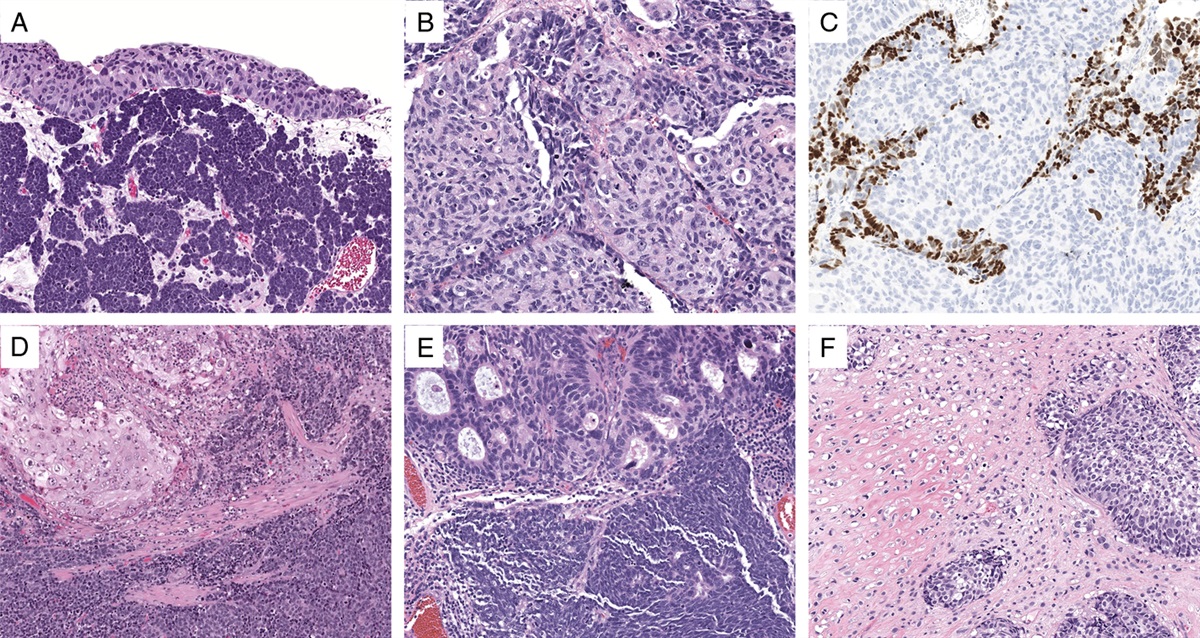
FIGURE 5: Malignant testicular sex cord–stromal tumor with EWSR1::ATF1 (inflammatory and nested testicular sex–cord tumor). (A) This neoplasm metastasized to the lung. (B) The tumor exhibits a nested architecture and a prominent lymphoplasmacytic infiltrate.
A subset of malignant Sertoli cell tumors exhibit predominantly solid growth with nests of tumor cells with granular eosinophilic to clear vacuolated cytoplasm; prominent inflammatory infiltrates with abundant lymphocytes and plasma cells are also typically present (Fig. 6).41 These morphologic features overlap with those of seminoma and these cases have been often seen in consultation by genitourinary pathologists with a prior diagnosis of seminoma or with a differential diagnosis that includes seminoma.41 Compared with seminoma, these TSCSTs have smaller nucleoli, lower mitotic activity (usually, but not always, <5 mitoses per 10 high-power fields), and a more heterogeneous inflammatory infiltrate that may include combinations of lymphocytes, plasma cells, neutrophils, and eosinophils.16,41 Immunohistochemistry demonstrates that they express sex-cord markers such as SF1 instead of SALL4 and OCT3/4.16 Recent analyses with massively parallel sequencing and fluorescence in-situ hybridization showed that these malignant TSCSTs harbor EWSR1::ATF1 fusions like the ones seen in clear cell sarcoma of soft parts and angiomatoid fibrous histiocytoma, among other tumor types.16 The fusion product contains exons 1-6/7 of EWSR1 and exons 4-7 of ATF1, coding for a protein that has the transactivation domain of EWSR1 and the DNA binding domain of ATF1.16 Given the distinct morphologic, molecular, and clinical features of these neoplasms, it has been suggested that they should be considered a separate entity (inflammatory and nested testicular sex–cord tumor).16 Importantly, RPLND should probably be considered for all patients presenting with these tumors.
 FIGURE 6:
FIGURE 6: Molecular alterations in Sertoli cell tumor, NOS. NOS indicates not otherwise specified. Figure created with BioRender.com.
Large Cell Calcifying Sertoli Cell TumorLCCSCT may occur sporadically or in the context of the Carney complex (~10% to 40%).4,6,11,21,48 Rare cases have also been described in patients with neurofibromatosis 1 and Peutz-Jeghers syndrome, although it is possible that some of the latter represent examples of intratubular large cell hyalinizing Sertoli cell neoplasia misinterpreted as LCCSCT.11 Syndromic neoplasms usually affect younger patients, are more frequently bilateral or multifocal, and mostly (but not exclusively) benign.49
Microscopically, LCCSCTs are composed of polygonal cells with abundant bright eosinophilic cytoplasm that resemble those of Leydig cell tumors, but with smaller nucleoli and no Reinke crystals.4,21,49 Multiple growth patterns have been described, including solid nests, solid sheets, trabeculae, and cords. The stroma is characteristically myxoid and edematous, with scattered laminated “mulberry-like” calcifications and a variably prominent neutrophilic infiltrate.4 Lymphocytes and lymphoid aggregates are also frequently seen, especially at the periphery of the tumor.11 Kratzer and colleagues found that malignant LCCSCTs often exhibit 2 or more of the following findings: size >4 cm, nuclear pleomorphism, necrosis, >3 mitoses per 10 high-power fields, vascular invasion, cytologic atypia, and invasion of extratesticular structures.11,49 However, as with other types of TSCSTs, there are neoplasms with worrisome histopathologic features that do not metastasize and, conversely, neoplasms without aggressive features that behave in a malignant fashion.11
Like other neoplasms associated with the Carney complex, both syndromic and sporadic examples of LCCST harbor PRKAR1A variants.50–52PRKAR1A codes for a regulatory subunit of the protein kinase A complex, which has an inhibitory effect on the catalytic subunits. Under physiologic conditions, cAMP produced by adenylate cyclase binds to the regulatory subunits and promotes their dissociation from the catalytic subunits, thereby releasing their inhibitory effect. Loss-of-function PRKAR1A mutations result in constitutive activation of the catalytic subunits and upregulation of PKA and MAPK signaling.53 Initial analyses of a limited number of cases with Sanger sequencing suggested that PRKAR1A variants were present in a heterozygous state; however, a more recent study with massively parallel sequencing has shown that loss-of-heterozygosity is likely present in most LCCSCT.50–52 In primary indolent tumors, PRKAR1A alterations are usually the only molecular finding, whereas metastasizing tumors often harbor additional genomic alterations, including chromosomal imbalances and further mutations.52 From a practical perspective, more than 90% of LCCSCTs, including both syndromic and sporadic cases, demonstrate loss of expression of PRKAR1A protein, which can be easily assessed with immunohistochemistry (Fig. 7).13
 FIGURE 7:
FIGURE 7: PRKAR1A immunohistochemistry in LCCSCT. (A) Aggressive LCCST with minimal calcifications and relatively subtle neutrophilic infiltrate. (B) The tumor demonstrates the loss of PRKAR1A expression (red chromogen). Most sporadic and syndromic LCCSCTs demonstrate the loss of PRKAR1A, a finding that may be particularly helpful in cases without typical histologic features. LCCST indicates large cell calcifying Sertoli cell tumor.
GRANULOSA CELL TUMORS OF THE TESTISGranulosa cell tumors of the testis are divided into adult-type (AGCT) and juvenile-type (JGCT).4,6,9,21,54 Both entities are defined based on their morphologic resemblance to ovarian counterparts, although they have significant clinical and molecular differences.55–57
AGCTs comprise tumors with a somewhat wide morphologic and clinical spectrum. AGCTs are composed of epithelioid and/or spindle cells with nuclei that typically display intranuclear grooves (“coffee-bean” shaped). Growth patterns are quite variable, including solid sheets, nests, microfollicles (Call-Exner bodies), and trabeculae. AGCT is largely a postpubertal neoplasm, with a mean age at diagnosis of 42 years.58 The most common presentation is as a painless testicular mass, but ~10% exhibit clinical findings secondary to the production of sex hormones.58 Despite the morphologic similarities between testicular and ovarian AGCTs, their underlying oncogenic mechanisms appear to be different. More specifically, the FOXL2 p.C134W variant, present in >90% of ovarian AGCTs, is only rarely identified in testicular cases.55,57,59–61 The genomic landscape of AGCTs appears to be heterogeneous and the only recurrent finding has been the loss of the long arm of chromosome 22 (~70%); however, this alteration is highly prevalent across many tumor types and likely represents a random recurrent event not associated with oncogenesis.55 Hence, testicular AGCT is a diagnostic category that seems to comprise a heterogeneous group of lesions that may represent different entities, including rare neoplasms with FOXL2 p.C134W.
JGCT is mostly an infantile neoplasm, with ~90% of cases occurring in patients of up to 6 months of age in some series (including a significant subset present at birth).9,58 JGCTs are sometimes associated with undescended testes or arise in dysgenetic gonads.9 Their morphologic features closely resemble those of ovarian JGCTs, with typical examples exhibiting small round tumor cells arranged in nests and follicles that contain eosinophilic or basophilic secretions. The stroma is prominent and fibrous or fibromyxoid, and mitotic activity is usually brisk.4,21 Testicular JGCTs are invariably indolent and do not recur after complete surgical excision.9,58 In contrast, ovarian JGCTs affect young prepubertal children with a median age of 7 to 8 years but may also arise in postpubertal women, and a subset behaves aggressively.62,63 Moreover, a significant proportion of ovarian JGCTs is associated with precocious pseudopuberty, whereas most testicular JGCTs are hormonally quiescent.62 Molecular pathogenesis of ovarian JGCTs is characterized by the presence of GNAS and AKT1 variants.64–66GNAS variants are typically hotspot mutations that decrease the GTPase function of the protein, keeping it in a constitutively activated (ie, GTP-bound) state.67 The AKT1 variants described in ovarian JGCTs include internal tandem duplications and mutations that affect conserved residues within the pleckstrin-homology domain of the protein, leading to its translocation to the plasma membrane and subsequent activation by kinases.65 Recent studies of a series of testicular JGCTs demonstrated that they lack pathogenic GNAS and AKT1 variants; instead, monosomy 10 is found in ~60% of cases, possibly reflecting a loss-of-heterozygosity event for a yet unidentified tumor suppressor.56
MYOID GONADAL STROMAL TUMOR AND SEX CORD–STROMAL TUMORS WITH PROMINENT OR PURE SPINDLE CELL COMPONENTSSeveral TSCST types are characterized by the presence of a prominent or pure spindle cell component, including AGCT, tumors with mixed sex cord, and stromal components (eg, “Sertoli-stromal” cell tumor, unclassified TSCSTs, mixed TSCSTs containing entrapped germ cells), fibroma/thecoma, and myoid gonadal stromal tumor.4,6,54,68–72 The latter represents a new entity in the World Health Organization (WHO) classification of urinary and male genital tumors and is somewhat arbitrarily defined as a pure stromal tumor that coexpresses SMA and S100.6 Except for a subset of AGCT, unclassified TSCST, and (possibly) some mixed TSCSTs, the remaining tumor types mentioned above are clinically indolent, being cured by complete surgical resection. Morphologic and immunophenotypic differences between these tumor types have been used to justify their classification as separate entities. As mentioned above, some of these neoplasms (AGCT, fibroma/thecoma) have been interpreted as testicular counterparts of ovarian tumors with similar morphology, whereas others (myoid gonadal stromal tumor, “Sertoli-stromal” cell tumors) seem to occur only in the testis.68,69,71,72 Interestingly, myoid gonadal stromal tumors, “Sertoli-stromal” cell tumors73, some testicular AGCTs, and a subset of unclassified TSCSTs with prominent spindle cell components harbor numerous recurrent chromosomal gains, suggestive of a shift in ploidy.17,18,55 Moreover, recurrent mutations have not been identified in any of these tumor types.17,18 Hence, it appears that these morphologically heterogeneous spindle cell-rich neoplasms that are currently classified into different entities harbor similar genomic alterations and may be part of the same biological spectrum. From a practical perspective, it is important to identify rare tumors within this group that exhibit multiple worrisome histopathologic features, indicating a significant risk of malignancy. These aggressive neoplasms often contain at least focal sex–cord components and can be categorized as adult granulosa cell tumors, mixed TSCSTs, or unclassified TSCST (ie, TSCST, NOS) according to their morphologic features. In contrast, based on limited published data, pure spindle cell TSCSTs seem to be invariably benign. Bland pure spindle cell TSCSTs that coexpress SMA and S100 can be classified as myoid gonadal stromal tumors (per WHO definition), whereas the remaining ones are generally interpreted as tumors within the fibroma/thecoma family or as TSCST, NOS. Whether several subtypes of TSCSTs with pure or predominant spindle cell components included in the latest WHO classification truly represent distinct entities or not remains an open question, requiring further investigation.
CONCLUSIONSTSCSTs comprise a group of benign and malignant neoplasms that recapitulate the phenotype of noneoplastic sex cord and stromal elements of the gonads. During embryogenesis, the primitive undifferentiated sex cords originate from invaginations of the celomic epithelium. In the primitive sex cords of XY embryos, a highly coordinated sequence of expression of transcription factors that include WT1 (KTS+ isoform), SRY, and SOX9 as major players will result in the differentiation of Sertoli cells, which are the source of anti-mullerian hormone, being therefore required for normal testicular development.74–77 In contrast, the stromal cells of the testis are thought to comprise subsets that derive from sex-cord precursors, the mesoderm of the mesonephros, and/or the neural crest.78–80 In the absence of specific biomarkers, the different types of TSCSTs have been historically subclassified based on their morphologic resemblance to nonneoplastic sex cord and stromal cells of the testis and ovary, with Leydig and Sertoli cell tumors being the most common histologic subtypes. Currently, TSCSTs are thought to originate from normal sex-cord or stromal elements of the testis that undergo neoplastic transformation, resulting in somewhat rigid categories that may not accommodate neoplasms with intermediate or mixed phenotypes. Such tumors are typically grouped under poorly defined categories (mixed TSCST and TSCST NOS) that are biologically and molecularly heterogeneous.18 Moreover, a classification that is based solely on morphology may result in excessive splitting, with the creation of spurious entities that may represent phenotypic variants of a single tumor type. Instead, a classification that integrates morphologic, immunophenotypic, and molecular findings may better reflect the biological and clinical features of TSCSTs. Similar classifications have been adopted for multiple tumor types across organ systems, demonstrating benefits for prognostication, clinical management, and treatment.
Recently, molecular analyses have described genetic alterations associated with oncogenesis and biological progression in several subtypes of TSCSTs. For instance, clonal gain-of-function CTNNB1 variants or loss-of-function APC variants are almost invariably found in Sertoli cell tumors, NOS without aggressive histopathologic or clinical features.42,44 In this tumor type, diffuse nuclear expression of beta-catenin can be used as a surrogate marker of the underlying genomic alterations for diagnostic purposes. The same applies to PRKAR1A immunohistochemistry for the diagnosis of LCCSCT, especially when these tumors lack the characteristic morphologic features.13 Other biomarkers, such as FH and MDM2 immunohistochemistry may be useful to identify Leydig cell tumors with high risk of malignant behavior.14,27 Moreover, molecular assessment of TSCSTs has identified potential new entities, such as malignant sex–cord tumors driven by EWSR1::ATF1 fusion (“inflammatory and nested testicular sex–cord tumor”), and suggested that some currently accepted entities (eg, myoid gonadal stromal tumor) may need to be reconsidered.16
In conclusion, the field of TSCSTs is evolving rapidly; in the coming years, further assessment of the genomic, epigenomic, and proteomic landscape of these neoplasms may help us better understand their oncogenesis and biologic features, leading to a more accurate and clinically useful classification. Moreover, refinement and validation of multiparametric risk assessment systems that incorporate clinical, histopathologic, and molecular data may help improve the identification of patients with testis-confined disease that require closer follow-up and a more aggressive therapeutic approach, including upfront retroperitoneal lymph node dissection.
REFERENCES 1. Dilworth JP, Farrow GM, Oesterling JE. Non-germ cell tumors of testis. Urology. 1991;37:399–417. 2. Cecchetto G, Alaggio R, Bisogno G, et al. Sex cord-stromal tumors of the testis in children. A clinicopathologic report from the Italian TREP project. J Pediatr Surg. 2010;45:1868–1873. 3. Thomas JC, Ross JH, Kay R. Stromal testis tumors in children: a report from the prepubertal testis tumor registry. J Urol. 2001;166:2338–2340. 4. Ulbright TM, Kao C-S, Williamson SR, et al. Tumors and Tumor-Like Lesions of the Testis and Adjacent Tissues. American Registry of Pathology; 2022. 5. Cheville JC. Classification and pathology of testicular germ cell and sex cord-stromal tumors. Urol Clin North Am. 1999;26:595–609. 6. WHO Classification of Tumours Editorial Board. Urinary and Male Genital Tumours, 5th ed. International Agency for Research on Cancer; 2022. 7. Nicolai N, Necchi A, Raggi D, et al. Clinical outcome in testicular sex cord stromal tumors: testis sparing vs. radical orchiectomy and management of advanced disease. Urology. 2015;85:402–406. 8. Kathrins M, Kolon TF. Malignancy in disorders of sex development. Transl Androl Urol. 2016;5:794–798. 9. Kao C-S, Cornejo KM, Ulbright TM, et al. Juvenile granulosa cell tumors of the testis: a clinicopathologic study of 70 cases with emphasis on its wide morphologic spectrum. Am J Surg Pathol. 2015;39:1159–1169. 10. Ulbright TM, Amin MB, Young RH. Intratubular large cell hyalinizing sertoli cell neoplasia of the testis: a report of 8 cases of a distinctive lesion of the Peutz-Jeghers syndrome. Am J Surg Pathol. 2007;31:827–835. 11. Al-Obaidy KI, Idrees MT, Abdulfatah E, et al. Large cell calcifying sertoli cell tumor: a clinicopathologic study of 18 cases with comprehensive review of the literature and reappraisal of prognostic features. Am J Surg Pathol. 2022;46:688–700. 12. Mosharafa AA, Foster RS, Bihrle R, et al. Does retroperitoneal lymph node dissection have a curative role for patients with sex cord-stromal testicular tumors? Cancer. 2003;98:753–757. 13. Anderson WJ, Gordetsky JB, Idrees MT, et al. Large cell calcifying Sertoli cell tumour: a contemporary multi-institutional case series highlighting the diagnostic utility of PRKAR1A immunohistochemistry. Histopathology. 2022;80:677–685. 14. Rizzo NM, Sholl LM, Idrees MT, et al. Comparative molecular analysis of testicular Leydig cell tumors de
留言 (0)