
記住我


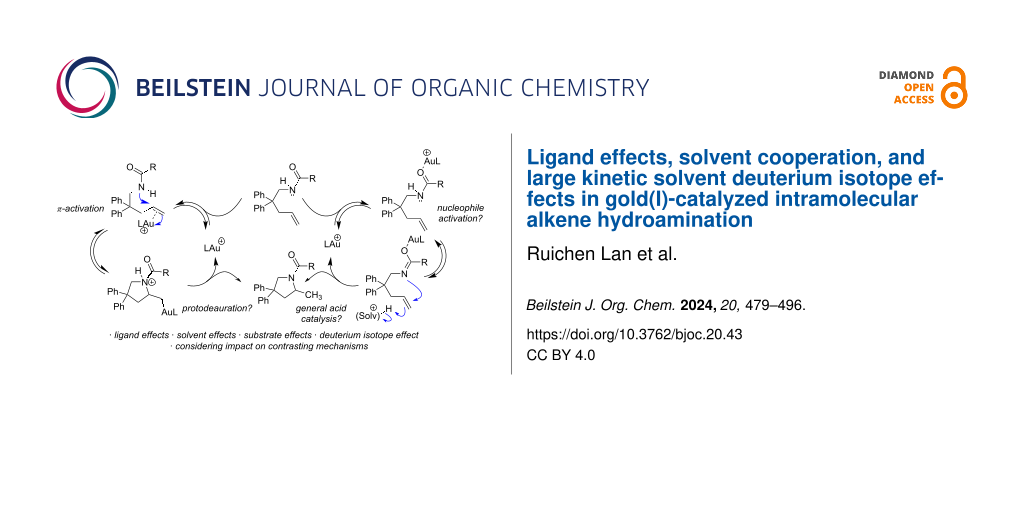
Since the seminal 1998 report by Teles et al. on the gold(I)-catalyzed addition of alcohols to alkynes , a multitude of gold-catalyzed reactions have been reported. Great successes in mechanistic analysis and synthetic methods have been achieved for allene and alkyne activation, while the activation of alkenes remains challenging. Advances in asymmetric catalysis , C–N and C–C functionalization reveal opportunities, but harsh conditions and limited substrate scope present problems. Intramolecular reactions almost invariably require geminal substitution or backbone heteroatoms, internal alkenes are often not tolerated, and intermolecular reactions require high temperatures which can lead to significant catalyst decomposition . This is usually addressed by employing bulky or strong donor ligands . Novel strategies tackle catalyst stability by changing the chloride scavenger or adding other coordinating moieties .
Hartwig et al. have argued that a Brønsted acid generated in situ from metal triflates may be the “real” catalyst promoting some alkene functionalizations . Therefore, the possibility of competing Brønsted acid catalysis in gold-catalyzed alkene functionalization remains a consideration , and while it is assumed that alkene activations follow the same prototypical mechanisms as allene and alkyne activations, that is (1) π-activation with nucleophilic attack followed by (2) protodeauration (Scheme 1), the depth of experimental mechanistic validation achieved for allenes and alkynes have not been reproduced with alkenes. In an important foundational study by Toste, the expected alkylgold intermediate from intramolecular alkene hydroamination was isolated, however, turnover protodeauration was not confirmed . Follow-up studies in our lab revealed that the alkylgold intermediate (2a, Scheme 1) reacts significantly slower than observed rates for catalytic hydroamination, suggesting it is not a viable intermediate in the catalytic cycle . It has been shown that C(sp2)-vinylgold intermediates (expected from allene/alkyne addition) are more reactive than the C(sp3)-alkylgold intermediates expected from alkene addition . Another study demonstrated the inefficiency of protodeauration in the presence of (albeit more basic) alkylamines . These studies cast doubt on protodeauration as the final step of alkene hydroamination, however, an alternative mechanistic model remains elusive.
![[1860-5397-20-43-i1]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-43-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Proposed mechanism and observation of alkylgold intermediates.
There are significant similarities between gold- and acid-catalyzed alkene additions, further confounding easy conclusions about the operative mechanism. Early gold-catalyzed alkene hydroaminations were shown to proceed with anti-selectivity and that was used as support for gold catalysis , but mechanism studies of triflic acid catalysis showed a preference for anti-selectivity as well . Despite similarities, control studies indicate meaningful differences in catalytic activity between gold and HOTf, however, they are not easily explained by simple either/or mechanisms .
Due in part to optimization challenges and in part to remaining gaps in characterizing structure–activity relationships for alkene hydroamination, we sought to obtain additional understanding by undertaking a 1H NMR spectroscopic kinetic survey of solvent, ligand, and substituent effects on the general reaction 1 → 3 (with a variety of N-protecting groups), to supplement known qualitative observations. We found that, (1) electron-withdrawing phosphines accelerate hydroamination, (2) reactions are faster with more Lewis basic urea substrates, (3) mixed solvents are uniquely able to enhance rates, with protic methanol and DCM identified as the best combination, and (4) kinetic isotope effects are variable depending on the concentration of protons in solution with small deuterium KIE’s at low concentration of deuterated species and large solvent KIE’s when performed in pure CH3OH versus CD3OD. Connections between catalyst activity and decomposition were made and a structurally interesting new bisphosphine–gold complex was isolated. Although our results do not provide conclusive evidence for or against turnover protodeauration, they indicate strong parallels to general acid catalysis. There is little doubt that gold is required for the transformation, but the combination of solvent and substrate effects suggest that instead of acting as a specific alkene activator, it may instead create generally acidic conditions that initiate cyclization . These observations are critical for informing future discussion and experiments related to this important reaction.
Results Ligand effectTo examine the catalytic activity of gold with ligands of different electronic properties, we used our recently developed series of bisbiphenylphosphine ligands, RP(o-biphenyl)2 (R = OPh, Ph, t-Bu) . When urea alkene 1a (0.1 M, CD2Cl2) was treated with 1 mol % LAuOTf, where L = PhOP(o-biphenyl)2 (4a), PhP(o-biphenyl)2 (4b), t-BuP(o-biphenyl)2 (4c), the fastest rate to form 3a was observed with the ligand with the greatest π-acceptor character and weakest donor character, namely L = PhOP(o-biphenyl)2 (Table 1) . Notable amounts of scatter and a narrow range of observed rates, however, reveal the differences to be relatively minor (Table 1, entry 1, krel(4a/4c) = 3.6). Nevertheless, each observed rate was outside of one standard deviation from the average of 3 trials, confirming the overall reactivity trend. In hindsight, the ligand effect here may be expected to be small since the bisbiphenyl scaffold contains 2 of 3 identical substituents. Nevertheless, these comparisons confirm the correlation of faster rate with more electron-withdrawing ligand. Experiments with other substrates display consistent results (further discussion below, Table 6, catalyst 4a reacts faster than 4c in the reaction of carbamate 1b → 3b).
Table 1: Relative rates of 1a hydroamination with gold phosphine triflate catalysts (average of 3 trials each).
![[Graphic 1]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-43-i3.svg?max-width=637&scale=1.0) Entry
R
105∙kobs/s−1
krel
1
PhO (4a)
206 ± 37
3.6
2
Ph (4b)
106 ± 13
1.8
3
t-Bu (4c)
58 ± 27
1
Solvent effect
Entry
R
105∙kobs/s−1
krel
1
PhO (4a)
206 ± 37
3.6
2
Ph (4b)
106 ± 13
1.8
3
t-Bu (4c)
58 ± 27
1
Solvent effect
For a screen of solvent effect on the rate of 1a hydroamination to 3a, commercially available ((acetonitrile)[(2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate (5) was used as catalyst (Figure 1), where gold is supported by the ligand commonly known as “Johnphos” or, henceforth, “JPhos”. Interestingly, there are minor differences in observed rate (first order fit in alkene decay) between THF (ε = 7.6, polarity index = 4.0), methylene chloride (ε = 8.9, polarity index = 3.1), and methanol (ε = 32.7, polarity index = 5.1) despite the large differences in solvent polarity (Table 2, entries 3, 4, and 5; rates in THF and MeOH are only 1–2 times faster than those in CD2Cl2). However, this did not hold true uniformly. When a commercially available electron-acceptor ligand known as “Jackiephos” (bis(3,5-bis(trifluoromethyl)phenyl)(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine was used as the AuNTf2 salt (6a), in CD2Cl2, the reaction rate from 1a → 3a was too fast to measure (reaction complete in t = 5 min, a >35 fold increase compared to MeOH, see Supporting Information File 1, Figure S12). Furthermore, the reaction rate with JackiephosAuNTf2 in pure MeOH was slightly slower than that of JPhosAuOTf in MeOH, suggesting a polar protic solvent minimizes ligand effects. Acetonitrile and deuterated methanol significantly decelerate the reaction (Table 2, entries 6 and 7; 10–20 times slower than in CD2Cl2), while a curious cooperative acceleration was observed when methanol or methanol-d4 were used in combination with methylene chloride (Table 2, entries 1 and 2; 4–22-fold increase in rate compared to CD2Cl2). We were unable to find any clear correlation between rate and a variety of solvent parameters; the success of mixed solvents suggests many factors are at play. Throughout our studies, we used CH2Cl2 and CD2Cl2 interchangeably; there is a slight difference in rate between the two (Table 2, entry 5, krel = 1.4 CH2Cl2 versus CD2Cl2). The slightly faster reaction in CH2Cl2 we believe can be attributable to different levels of H2O contaminant.
![[1860-5397-20-43-1]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-20-43-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: First order alkene decay for urea alkene 1a (0.05 M) hydroamination with [JPhosAu(NCCH3)]SbF6 (5, 2.5 mol %) in various solvents.
Table 2: Relative rates of hydroamination with [JPhosAu(NCCH3)]SbF6 (2.5 mol %) and urea alkene 1a (0.05 M) in various solvents.
![[Graphic 2]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-43-i4.svg?max-width=637&scale=1.0) Entry
Solvent
105∙kobs/s−1
krel
1
CD2Cl2/
Entry
Solvent
105∙kobs/s−1
krel
1
CD2Cl2/
aAdditional trials with JPhosAuOTf.
To quantify the cooperative accelerating effect of CH3OH combined with DCM in the reaction of 1a → 3a, we performed rate studies with titrated amounts of MeOH (Figure 2). In CD2Cl2 the rate of 1a disappearance increases steadily with each increase of MeOH (from 0–55 μL, or 0.18 M–1.94 M). We expect rates to plateau and then decrease, since the rates in pure MeOH are slower (see above, Table 2, entry 4). We were not able to determine the maximum impact of added MeOH, because the rate of cyclization became too fast to be detectable by 1H NMR kinetics (t < 5 minutes). A LN-LN plot of MeOH concentration versus observed rate gives a slope of 0.7, indicating less than first order dependence on MeOH.
![[1860-5397-20-43-2]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-20-43-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Cooperative effect of mixed CD2Cl2/MeOH on alkene 1a → 3a conversion with catalyst 5 (2.5 mol %). Error bars are from linear least squares analysis of raw data plots.
A similar acceleration in 1a hydroamination is seen when water additive is combined with DCM solvent, but unexpectedly, subtle differences were observed depending upon the identity of the catalyst (Table 3, Figure 3). Early experiments used JPhosAuOTf (synthesized in our lab) and water as co-solvent, but the conditions were modified to use readily accessible commercial [JPhosAu(NCCH3)]SbF6 (5) and MeOH, since water posed miscibility problems at higher concentrations. Titrating increasing amounts of water into reactions with JPhosAuOTf (4d) led to an increase in rate, similar to that seen with MeOH and catalyst 5, albeit smaller in magnitude (Table 3, entries 1–4 show the increasing rate with increasing water up to a 5.3-fold increase at 3.2 M water, while entries 5–8 show the increasing rate with increasing methanol up to a 14.6-fold increase at only 1.9 M methanol). In contrast, titrating increasing amounts of water into reactions of 1a with [JPhosAu(NCCH3)]SbF6 (5) led to an initial boost that quickly plateaued (Table 3, entry 5 compared to entries 9–11). Furthermore, when water was added to the reaction catalyzed by JPhosAuOTf (4d) in MeOH, the reaction slowed with increasing amounts of water (Table 3, entries 12–14) . The same decelerating effect of water in MeOH solvent was seen with [JPhosAu(NCCH3)]SbF6 (5) (Table 3, entries 15–17).
Table 3: Impact of titrated water or methanol into CD2Cl2 or CH3OH.
![[Graphic 3]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-43-i5.svg?max-width=637&scale=1.0) Cat.
Additive
kobs∙105/s−1
Solvent CD2Cl2
1
4d
0 M water
47 ± 3
2
4d
0.16 M water
67.4 ± 0.6
3
4d
0.8 M water
121 ± 9
4
4d
3.2 M water
249 ± 13
5
5
0 M
37 ± 1
6
5
0.18 M MeOH
97 ± 3
7
5
0.9 M MeOH
381 ± 6
8
5
1.9 M MeOH
539 ± 30
9
5
0.4 M water
95 ± 3
10
5
2 M water
110 ± 3
11
5
4.4 M water
104 ± 3
Solvent MeOH
12
4d
0 M water
48.2 ± 0.8
13
4d
2 M water
19.5 ± 0.2
14
4d
3.2 M water
11.1 ± 0.4
15
5
0 M water
50 ± 1
16
5
2 M water
11.5 ± 0.3
17
5a
6 M water
6.5 ± 0.4
Cat.
Additive
kobs∙105/s−1
Solvent CD2Cl2
1
4d
0 M water
47 ± 3
2
4d
0.16 M water
67.4 ± 0.6
3
4d
0.8 M water
121 ± 9
4
4d
3.2 M water
249 ± 13
5
5
0 M
37 ± 1
6
5
0.18 M MeOH
97 ± 3
7
5
0.9 M MeOH
381 ± 6
8
5
1.9 M MeOH
539 ± 30
9
5
0.4 M water
95 ± 3
10
5
2 M water
110 ± 3
11
5
4.4 M water
104 ± 3
Solvent MeOH
12
4d
0 M water
48.2 ± 0.8
13
4d
2 M water
19.5 ± 0.2
14
4d
3.2 M water
11.1 ± 0.4
15
5
0 M water
50 ± 1
16
5
2 M water
11.5 ± 0.3
17
5a
6 M water
6.5 ± 0.4
aAt 75 μL water in CH3OH precipitates begin to form and the sample is turbid.
![[1860-5397-20-43-3]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-20-43-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Different additive impact on rate of 1a → 3a depending upon catalyst and co-solvent. The data for JPhosAu(NCCH3)SbF6 with MeOH in DCM (▲) is reproduced in Figure 2. (The reaction with catalyst 5 and water in MeOH is not shown but displays similar inhibition). Error bars are from linear least squares analysis of raw data plots; where not visible they are smaller than the icon for the data point.
Substrate effectThe rate of hydroamination to cyclized 3a–c was measured for three substrates, tert-butylurea 1a, tert-butyl carbamate 1b, and benzamide 1c (Table 4). Under standard conditions (2.5 mol % [JPhosAu(NCCH3)]SbF6, 0.05 M alkene in DCM) benzamide and carbamate hydroamination were too slow to measure, so the reactions were done with 55 μL MeOH promoter but still only an estimated rate constant was obtained for 1c (14% conversion after 24 h, estimated t1/2 = 96 h, kobs = 1.4 × 10−6 s−1). With 55 μL MeOH in DCM, the relative rates for each substrate are 3,850:50:1 with urea 1a > carbamate 1b > benzamide 1c. The analogous toluene sulfonamide substrate 1d did not react on measurable timescales at room temperature (no product with up to 10 mol % JPhosAu(NCCH3)SbF6 in CD2Cl2 after 48 hours with and without added CH3OH) despite common use of sulfonamides in alkene hydroamination reports, albeit at higher temperatures.
Table 4: Relative rates of hydroamination with different protecting groups on nitrogen; (1a–c, 0.05 M) with catalyst 5 (2.5 mol %) in DCM and 55 μL CH3OH promoter (1.9 M).
![[Graphic 4]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-43-i6.svg?max-width=637&scale=1.0) Entry
Substrate
kobs∙105/s−1
1
1a/CD2Cl2
539 ± 30
2
1b/CH2Cl2
6.9 ± 0.2
3
1c/CH2Cl2
≤0.14
4
1d/CD2Cl2
n.d.*
Entry
Substrate
kobs∙105/s−1
1
1a/CD2Cl2
539 ± 30
2
1b/CH2Cl2
6.9 ± 0.2
3
1c/CH2Cl2
≤0.14
4
1d/CD2Cl2
n.d.*
*No reaction with 10 mol % catalyst 5.
Based on our ligand survey above, we proposed to improve rates of cyclization with slower reacting substrates by identifying a more Lewis acidic gold. To determine whether benzamide (1c) cyclization could be made efficient with appropriate combination of ligand and MeOH we surveyed rates with (PhO)P(o-biphenyl)2AuOTf (4a) and JackiephosAuNTf2 (6a, Table 5). Benzamide rates remain slow but gains can be achieved – benzamide (1c) cyclization with Jackiephos and 55 μL MeOH promoter matches the rate of cyclization of carbamate (1b) with JPhosAu(NCCH3)SbF6 with only 5 μL MeOH promoter (see Supporting Information File 1, Figure S18). Increasing to 100 μL MeOH did not increase the benzamide cyclization rate any further (see Supporting Information File 1, Figure S19). This comparison reveals that the Jackiephos supported gold accelerates the reaction more than (PhO)P(o-biphenyl)2 does, although this may be in part an anion effect . The bis(trifluoromethyl)aryl substituent is expected to be more electron withdrawing than an o-biphenyl, so presumably Lewis acidity is boosted here.
Table 5: Benzamide (1c, 0.05 M) hydroamination with 2.5 mol % of two different catalysts in CH2Cl2 with 55 μL CH3OH.
![[Graphic 5]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-43-i7.svg?max-width=637&scale=1.0) Entry
Catalyst
105∙kobs/s−1
1
4a
0.37 ± 0.02
2
6a
1.02 ± 0.09
Observations on ligand effect and decomposition
Entry
Catalyst
105∙kobs/s−1
1
4a
0.37 ± 0.02
2
6a
1.02 ± 0.09
Observations on ligand effect and decomposition
Ligand effects on rates of hydroamination are amplified with slower reacting substrates and lower amounts of MeOH (higher relative amounts of bulk solvent methylene chloride). With the carbamate substrate 1b, the rate of cyclization is 10× faster with PhO(o-biphenyl)2PAuOTf (4a) compared to t-Bu(o-biphenyl)2PAuOTf (4c) when no MeOH additive is used, and only ≈2× faster with 5 and 25 μL of MeOH (see Table 6). This contrasts the 3.6 fold difference in rate depending upon catalyst identity (k(4a/4c)) for urea substrate 1a → 3a and no MeOH (see Table 1).
Table 6: Influence of increasing MeOH and catalyst (2.5 mol %) on carbamate 1b (0.05 M in CH2Cl2) reactivity.
![[Graphic 6]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-43-i8.svg?max-width=637&scale=1.0) Entry
Additive
Cat.
105∙kobs/s−1
1
none
4aa
1.981 ± 0.007
2
5 μL MeOH
4aa
17.01 ± 0.08
3
25 μL MeOH
4aa
21.7 ± 0.1
4
none
4c
0.205 ± 0.009
5
5 μL MeOH
4c
8.58 ± 0.01
6
25 μL MeOH
4c
12.68 ± 0.07
Entry
Additive
Cat.
105∙kobs/s−1
1
none
4aa
1.981 ± 0.007
2
5 μL MeOH
4aa
17.01 ± 0.08
3
25 μL MeOH
4aa
21.7 ± 0.1
4
none
4c
0.205 ± 0.009
5
5 μL MeOH
4c
8.58 ± 0.01
6
25 μL MeOH
4c
12.68 ± 0.07
aDecomposition observed.
Hydroaminations with slower reacting substrates revealed another important factor. 31P NMR spectra during the cyclization of carbamate 1b indicated significant amounts of catalyst decomposition when PhOP(o-biphenyl)2AuOTf (4a) was used, however, the appearance of decomposed catalyst was not sufficiently detrimental to halt the reaction altogether. Catalyst decomposition is typically detected in two ways, either by the appearance of diagnostic [L–Au–L]+ in the 31P NMR spectrum, or by observation of a kinetic plateau in the reaction rate . In the reactions monitored here, the observed extent of decomposition is dependent on ligand and substrate. For example, in the cyclization of the less reactive carbamate substrate, with catalyst 4c the decomposed product [L–Au–L]+ was not observed on the timescale of the reaction, whereas with catalyst 4a, the decomposed product was observed. Also noteworthy, despite the higher amount of decomposition, ligand 4a still created a more effective catalyst system (twice as reactive, see Table 6). Furthermore, observed decomposition did not correlate with a reaction plateau; when in the presence of 5 μL MeOH, the first order plots of alkene decay retained linearity up to 80% conversion, and separate 31P NMR experiments indicated significant decomposition at about 50% conversion. Independently prepared [L–Au–L]+ has been shown by others to be inactive catalysts (L = Ph3P) and we confirmed this also with L = (t-Bu)2P(o-biphenyl). When the more reactive urea alkene 1a is examined, the reaction with 4a is efficient enough that reaction completion occurs prior to noticeable decomposition. In contrast, when a LAuNTf2 catalyst was prepared where L = tris(2,4-di-tert-butylphenyl)phosphite (6b), a supporting ligand that would be predicted to create more electrophilic gold due to its high π-acceptor properties, major decomposition was observed for the slower substrates (1b and 1c) and the fast urea (1a), indicating catalysts that are much more prone to decomposition, and preventing any meaningful determination of an actual ligand effect on reactivity (see Supporting Information File 1, Figures S25 and S26). These experiments indicate that with highly reactive catalysts, productive reactions can take place despite a drop in concentration of non-decomposed catalyst. The true reactivity of such catalysts may thus be anticipated to be much higher than the actual rates measured.
We noted recently that while the [Ph3P–Au–Ph3P]+ decomposition product makes a multitude of appearances in the literature, the corresponding complex has not be reported for JPhos, therefore we sought to independently prepare it. The structure ended up being unique and puzzling. When [LAu(NCCH3)]SbF6 was treated with an equivalent of free ligand, a complicated 31P NMR spectrum was acquired which we at first believed to be a result of erroneous choice of free ligand! Three signals were observed, a set of doublets at 107 and 69 ppm, with J = 275.5 Hz, and a singlet at 70.5 ppm (Figure 4c) . In all attempts to prepare bisphos 7a, the singlet and set of doublets were observed, and always in the same approximate ratio. X-ray crystallographic analysis shows C2 rotational symmetry and confirms the identity of complex 7a. Our preliminary hypothesis is that two conformations exist in solution, one of which is symmetrical (presenting as a singlet), one of which is not. A lack of symmetry in one conformation would mean that each phosphorous is magnetically inequivalent, and thus shows splitting to the other in the NMR spectrum. Although we observed reaction inhibition in a number of instances with [JPhosAu(NCCH3)]SbF6 we have never yet observed the formation of this byproduct. On the one hand, this is a testament to the high stability of gold supported by JPhos, on the other hand, it suggests as yet undetermined deactivation pathways. A similar 31P spectrum was obtained for the bisphoshine complex of Jackiephos, but in contrast no symmetrical singlet was observed (see Supporting Information File 1, Figure S23).
![[1860-5397-20-43-4]](https://www.beilstein-journals.org/bjoc/content/figures/1860-5397-20-43-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: (a) Schematic for synthesis of [L–Au–L]SbF6 where L = JPhos. (b) Perspective drawing of the cation in crystalline [Au(P(C4H9)2(C12H9))2](SbF6)CH2Cl2 where P are represented by dotted spheres, Au atoms are represented by cross-hatched spheres, and carbon and hydrogen atoms are represented by medium and small open spheres, respectively and all nonhydrogen atoms are labeled . (c) 31P NMR spectrum (161.98 MHz, CDCl3).
Kinetic deuterium isotope effectMonodeuterated methanol (CH3OD) was tested as an additive on the cyclization rate of alkene urea 1a with JPhosAu(NCCH3)SbF6 (5) in CH2Cl2 (Table 7, Figure 5). In this case an initial boost in reactivity was observed with 5 and 25 μL of CH3OD, followed by a drop-off in reactivity at 55 μL (even slower than in pure CH2Cl2). In all three experiments, rapid H/D exchange (t < 5 minutes) reduced the N–CH2 doublet (δ 3.84 ppm in CD2Cl2) to a singlet, indicating high incorporation of deuterium into the substrate and corresponding in situ generation of CH3OH. Furthermore, both NH signals appear absent in the 1H NMR spectrum (δ 3.93/3.66). Comparing the rates of reactivity to those with CH3OH as additive provides a range of KIE values, k(H/D) = 1.4 to 6.6. When the bulk solvent was changed from pure CH3OH to CD3OD a solvent isotope effect of 11.9 was measured.
Table 7: Influence of MeOH/MeOD on urea and carbamate reactivity.
![[Graphic 7]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-20-43-i9.svg?max-width=637&scale=1.0) Entry
Substrate/
Entry
Substrate/
留言 (0)