

記住我


Fanconi anaemia (FA) is a rare disorder characterised by progressive bone marrow failure (BMF), congenital dysmorphisms (including short stature, microcephaly, skin pigmentation abnormalities, thumb/radial ray malformations), cancer predisposition and hypersensitivity to DNA crosslinking agents (1, 2). FA presents with significant phenotypic variability, even between members of the same family with the same FA-associated mutation (3). A diagnosis of FA is often considered on the basis of BMF alongside congenital dysmorphisms. Diagnosis is usually confirmed by demonstration of in vitro chromosomal sensitivity to crosslinking agents, such as DEB (diepoxybutane) (4) or MMC (mitomycin C) (5). Following this, genetic testing can identify which FA-associated gene is mutated (6).
Mutation in twenty-two genes have been identified in FA: FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG/XRCC9, FANCI, FANCJ/BRIP1, FANCL, FANCM, FANCN/PALB2, FANCO/RAD51C, FANCP/SLX4, FANCQ/ERCC4/XPF, FANCR/RAD51, FANCS/BRCA1, FANCT/UBE2T, FANCU/XRCC2, FANCV/REV7/MAD2L2, and FANCW/RFWD3 (2). FANCH was identified as an FA gene but later found to be analogous to FANCA (7). The protein products of these genes interact to repair interstrand crosslinks (ICL) by regulating/directing nucleolytic incision, translesion synthesis, and homologous recombination (8). The FA core complex recognises ICLs at a stalled replication fork and ubiquitinates the FANCD2-FANCI (ID2) complex, which recruits downstream effectors of the FA pathway including FANCS/BRCA1. These mediate repair by nucleolytic incision, ICL unhooking, generation of double-strand breaks (DSB), and RAD51-dependent strand invasion and recombination (2, 8). Biallelic pathogenic mutations in these genes cause FA, hence inheritance follows an autosomal recessive pattern, with two exceptions: heterozygous pathogenic variants of FANCR/RAD51 cause autosomal dominant FA-R (9, 10) and hemizygous pathogenic variants of FANCB cause X-linked FA-B (11). FANCA, FANCC and FANCG mutations account for ~85% of FA cases (2), whereas FANCV and FANCW mutations have only been identified in one FA patient each (12–14).
Whilst biallelic (homozygous or compound heterozygous) mutations in FA-associated genes can result in FA, germline monoallelic mutations in many of these same genes may confer increased cancer risk. For example, biallelic mutations to BRCA2 underlie FA-D1, whereas monoallelic mutations are frequently observed in hereditary breast and ovarian cancer (HBOC), highlighting the link between FA and BRCA DNA repair pathways (15–17). Further, biallelic mutations in PALB2 and BRIP1 are associated with FA-N and FA-J respectively (18–20), but are associated with moderate HBOC risk in a heterozygous setting (21, 22). Despite identification of interaction between BRCA1 and known FA proteins (23), BRCA1 was not generally considered to be a canonical FA gene, as viable biallelic mutations affecting BRCA1 were not observed or expected. Mouse models demonstrated that most combinations of Brca1 biallelic mutations result in embryonic lethality and that one wild-type allele is required for development (24–27). However, there have now been a number of individual case reports of biallelic pathogenic BRCA1 mutations, many of whom have been identified as having a new form of Fanconi Anaemia – FA-S. This work conducted a scoping review in order to collate all reported cases of biallelic BRCA1 mutation and FA-S, to allow assimilation of clinical and genetic data on this rare condition.
MethodsA scoping review was performed by a standardized method. Two databases were used for the search, Medline and SCOPUS. In addition, a grey literature search was conducted using Google Scholar. On each, a standardised search string was used to search for human case reports of germline biallelic BRCA1 mutations:
● MEDLINE: (brca1 AND (homozygous OR biallelic OR (compound AND heterozygous))).ti.
● SCOPUS: Title((brca1 AND (homozygous OR biallelic OR (compound AND heterozygous))).
● Google Scholar: allintitle: brca1 homozygous OR biallelic OR “compound heterozygous”.
The following exclusion criteria were used:
1. Not reporting a human case of germline biallelic BRCA1 mutations due to:
a. No human case reported.
b. Involving a gene other than BRCA1.
c. Not germline biallelic mutations including:
i. Multiple BRCA1 mutations in cis rather than in trans.
ii. Non-germline mutations i.e. one or more somatic mutation sequenced in tumours.
2. Duplicates or other clear reasons for exclusion.
Data from included articles was collected in a standardised data collection table to ensure uniformity of data collection.
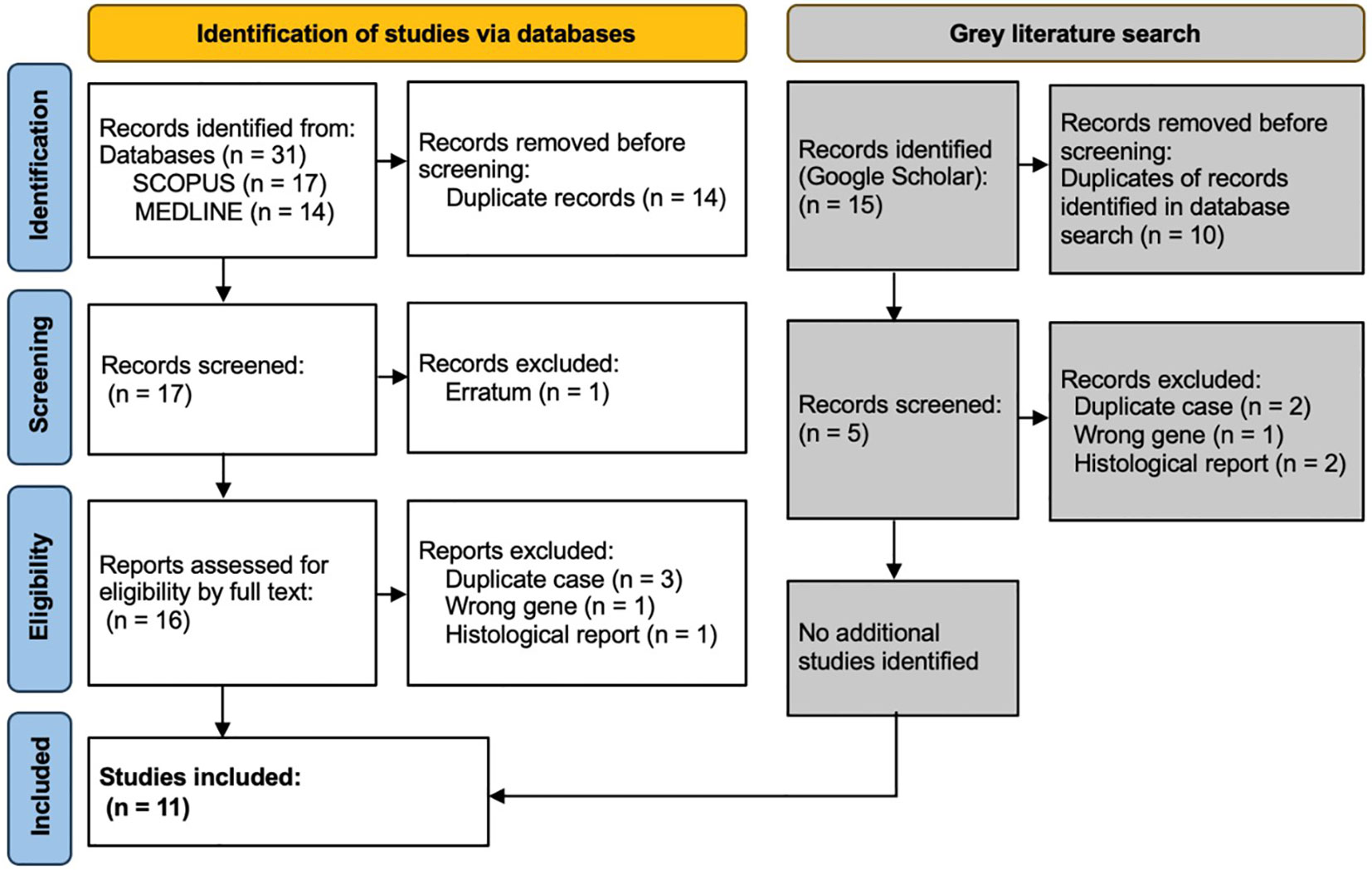
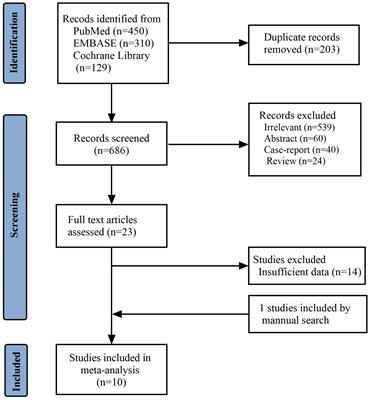
ResultsThe search on Medline gave 14 results, of which 3 were excluded: one was a discussion about tumour histology, one was an erratum (author name spelling error), and one was regarding a different gene (BARD1). SCOPUS provided 13 results and 4 secondary documents. All 13 of these were included within the 14 results already found via Medline, and the same 3 papers were excluded. Within the 4 secondary documents, 1 was the 14th paper found via Medline, and 3 were the initial case reports of cases later discussed in more detail within the previously identified papers and thus represented duplications of cases. The grey literature search using Google Scholar found no additional case reports. This resulted in 11 papers included in the detailed review (Figure 1).
Figure 1 PRISMA diagram showing selection of studies for inclusion in scoping review. Template and process from Page et al. (28).
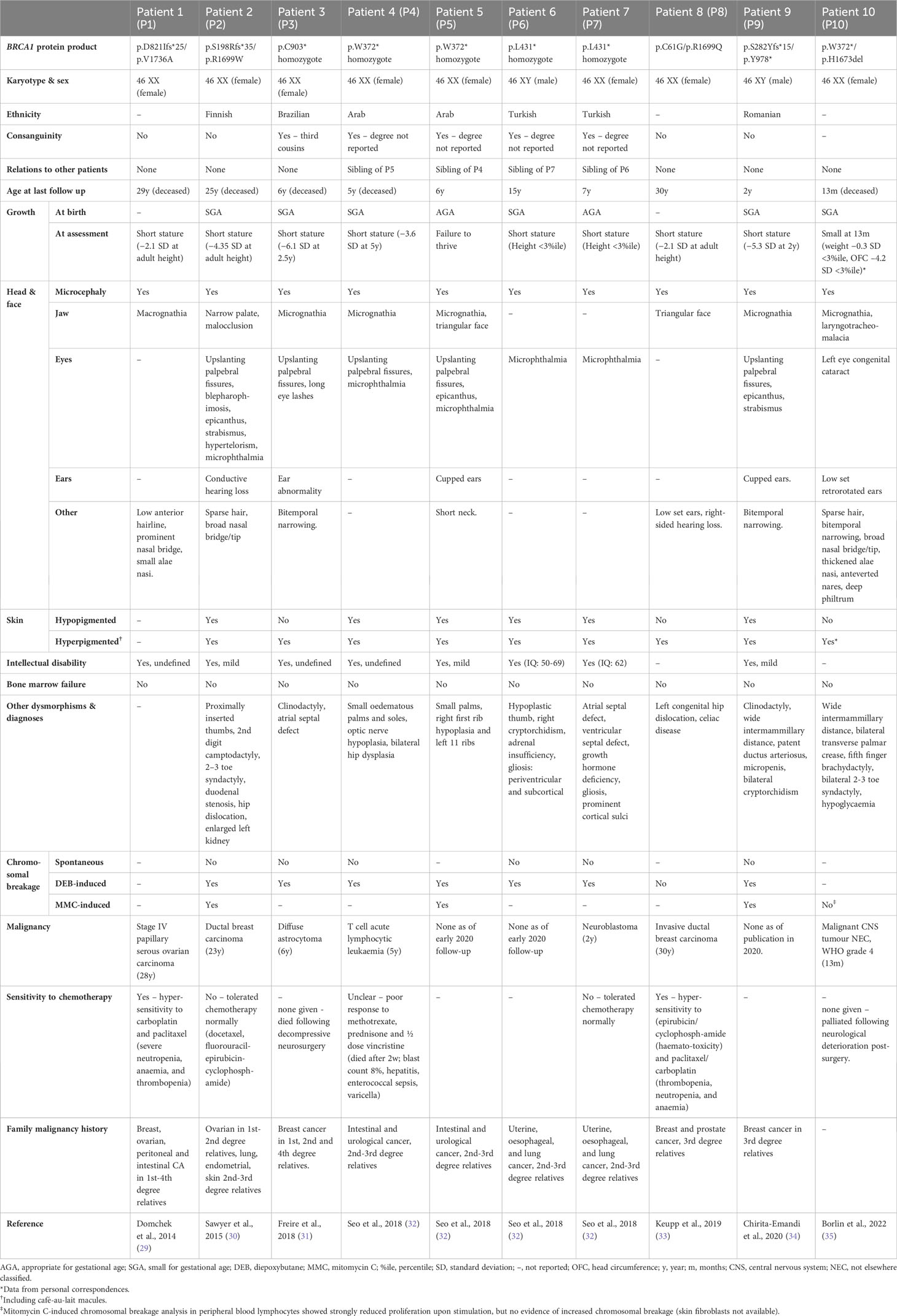
Fanconi anaemia type S patient cohortIn total, we have identified 12 cases of biallelic BRCA1 mutation, 10 in patients with an FA-S phenotype and 3 seemingly unaffected individuals (Tables 1, 2). A human with biallelic BRCA1 mutations was first reported in 1995, in a woman with breast cancer at age 32 and no other clinical features, described as homozygous for a high penetrance breast/ovarian cancer-associated mutation, but this report was subsequently found to be inaccurate (39). The case report suggested that homozygous BRCA1 c.2800delAA mutation did not increase cancer risk beyond heterozygosity. However, this report was later found to be inaccurate due to experimental error – there was an amplification bias during genotyping which led to relative excessive amplification of the mutant allele (40). Subsequent re-sequencing confirmed that both mutant and wild-type alleles were present (29).
Table 1 FA-S Cohort: clinical characteristics and genetic information.
Table 2 Individuals reported to have biallelic BRCA1 mutations without any FA-like phenotype: clinical characteristics and genetic information.
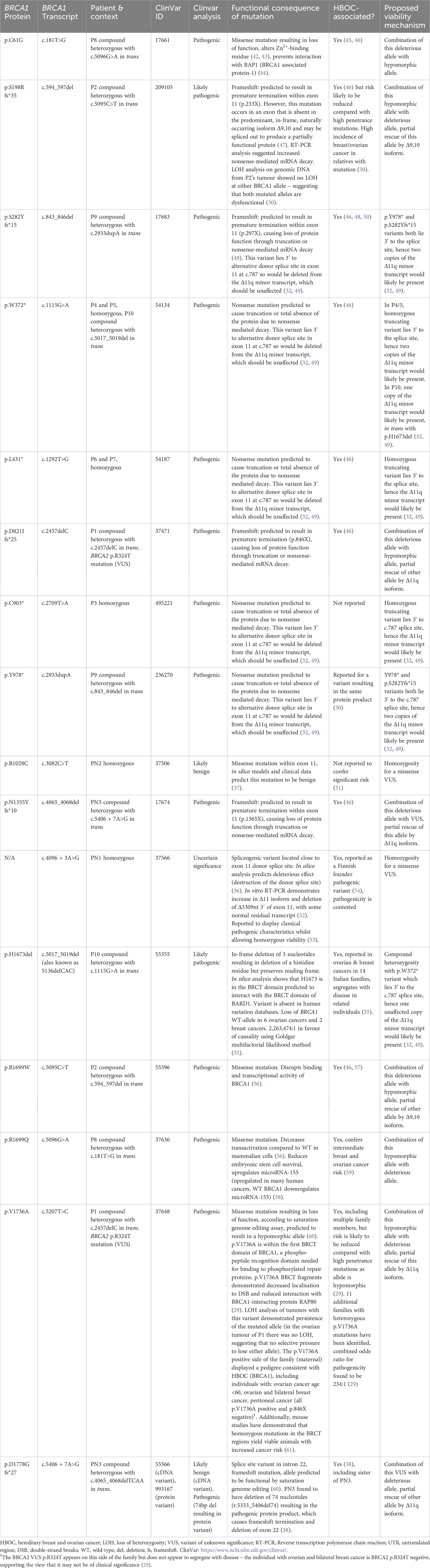
The first validated case of biallelic BRCA1 mutations was identified in 2013 by Domchek et al., in a woman with ovarian carcinoma at age 28 (P1, Table 1) (29). P1 is a BRCA1 compound heterozygote with a known deleterious mutation (p.D821Ifs*25) (41), in trans with a hypomorphic allele (p.V1736A). P1 also has a monoallelic BRCA2 mutation (p.R324T), which is a variant of unknown significance (VUS). Prior to publication the p.V1736A allele was classified as a VUS, but once identified in P1 it was thoroughly investigated genetically and biochemically to ascertain whether this patient represented a genuine case of biallelic pathogenic BRCA1 mutations. These investigations convincingly support the view that p.V1736A is a hypomorphic alteration affecting DNA repair (Table 3) (29). Interestingly, this patient was not originally suggested to have a form of FA, likely due to the absence of BMF. However, a number of clinical features differentiate P1 from a typical HBOC (BRCA1) phenotype. P1 experienced significant sensitivity to crosslinking chemotherapeutics (requiring discontinuation), and was reported to have short stature, microcephaly, facial dysmorphisms and intellectual disability. It is not known whether she had chromosome instability, as no breakage testing was undertaken prior to her death six months following cancer diagnosis.
Table 3 Detailed information regarding mutations found in individuals with biallelic BRCA1 mutations.
Sawyer et al. defined the FA-S subtype with the identification of P2 in 2015 (30). She presented with multiple congenital abnormalities including short stature, microcephaly, facial dysmorphisms, hypo- and hyper-pigmented skin lesions, proximally inserted thumbs (radial ray anomaly) and intellectual disability, as well as ductal breast carcinoma aged 23 (P2, Table 1). She had originally been suggested to have Dubowitz syndrome and was genotyped as part of an effort to identify Dubowitz-associated genes, during which she was found to have compound heterozygous BRCA1 mutations. A p.R1699W missense mutation, previously identified as pathogenic in HBOC (BRCA1) families (56), as well as a p.S198Rfs*35 mutation. DEB and MMC testing showed elevated chromosome breakage within diagnostic parameters for FA. Despite the chromosomal breakage results, she tolerated chemotherapy without signs of haematotoxicity. P2 has recently been discussed in a paper considering alternative genomic diagnoses for patients clinically diagnosed with Dubowitz syndrome but re-diagnosed with other disorders following genomic testing (62).
Freire et al. presented the first patient with homozygous pathogenic BRCA1 mutations – a girl (P3, Table 1) aged 2.5 years with similar dysmorphic phenotypic presentation to P1/P2 (31). P3 was found to have a homozygous nonsense BRCA1 mutation (p.C903*) resulting in premature termination within exon 11 and loss of key protein functional domains. Although this mutation has not been identified as an HBOC-associated variant, loss-of-function truncating mutations distal of this site have been reported as pathogenic, strongly suggesting pathogenicity (63). Cytogenic testing showed increased DEB-induced chromosomal breakage. Upon investigation, P3’s mother (p.C903* heterozygote) was found to have undifferentiated metastatic adenocarcinoma (31). P3 was not reported to have any malignancy in the original paper. Unfortunately, following publication, P3 developed a diffuse astrocytoma and died shortly following decompressive neurosurgery (64).
Following this, four patients (two pairs of siblings) with homozygous BRCA1 nonsense mutations were identified by Seo et al. – one pair with biallelic p.W372* mutations (P4 and P5, Table 1) and the other pair with biallelic p.L431* mutations (P6 and P7, Table 1) (32). These patients all presented with microcephaly, microphthalmia, abnormally pigmented skin lesions, intellectual disability and growth abnormalities (short stature or failure to thrive), as well as elevated chromosomal sensitivity to DEB and/or MMC. Interestingly, siblings P6/P7 presented with endocrine and neuroanatomical anomalies, which are not observed in any other FA-S patients to date and are not typical of FA. This may be a result of their particular BRCA1 mutation, or alternately caused by a separate undiagnosed condition. Endocrine abnormalities are common in FA, including growth hormone deficiency and hypothyroidism, but CNS anomalies present in only around 8% of FA cases (1). P4 and P7 developed childhood cancers, T-cell acute lymphocytic leukaemia (ALL) and neuroblastoma respectively. P7 responded normally to chemotherapy. However, P4 responded poorly to chemotherapy, even with a reduced-dosage regime. Although a haematotoxic response was not reported, P4 died soon after initiation of chemotherapy as a result of complex infections (Table 1). This might be suggestive of severe haematotoxicity and could represent a second patient in the cohort with sensitivity to crosslinking chemotherapeutics. As of an early 2020 follow-up, the status of patients P5-7 remains the same as at the time of publication (personal correspondence, 2021).
Three further patients have since been identified (P8, P9 and P10, Table 1). P8 was a compound heterozygote with two pathogenic BRCA1 mutations (33) – one high penetrance (p.C61G) (45) and one conferring intermediate risk (p. R1699Q) (59). P8 has congenital abnormalities consistent with previously identified FA-S patients, an early breast cancer (age 30), and had a haematotoxic response to crosslinking chemotherapeutics. Surprisingly, P8 did not exhibit DEB-induced chromosomal instability, and it is therefore disputable whether she should be considered a canonical FA-S patient. However, she is included here due to the burden of FA-S-associated clinical signs and lack of MMC testing. P8 does appear to have the mildest phenotype of FA-S patients (and is the oldest surviving FA-S patient to-date), but should certainly be distinguished from classical HBOC (BRCA1) patients on account of the congenital abnormalities, sensitivity to crosslinking chemotherapeutics, and presence of two pathogenic mutations in trans. P9 is the second male patient to be identified with FA-S (34), and is a compound heterozygote with two pathogenic truncating BRCA1 mutations in trans: p.S282Yfs*15 and p.Y978*. He presented with a phenotype similar to previous FA-S patients (growth restriction, multiple dysmorphic facial features and skin pigmentation) and exhibits chromosomal sensitivity to DEB and MMC, but no malignancy has been reported.
The most recently identified FA-S patient, P10, was a female compound heterozygote with a pathogenic mutation p.W372*, in trans with a p.H1673del mutation (35); the p.W372* mutation being the same as P4/5, and also reported as HBOC-associated (46). Genetic analysis strongly supports the view that p.H1673del is a pathogenic mutation, with association with an ovarian-predominant HBOC presentation in heterozygotes, loss of heterozygosity (LOH) in breast/ovarian cancers, and segregation with affected individuals in families (55). In silico modelling predicted an effect on BARD1 binding to the BRCT region, and multifactorial likelihood calculation gave a very high ratio in favour of causality (Table 3). P10 presented with multiple dysmorphic features including laryngotracheomalacia, abnormally pigmented skin lesions, distal skeletal abnormalities, and growth abnormalities. She developed a malignant CNS tumour (not elsewhere classified, WHO grade 4) at 13 months, and died 6 weeks later following a palliative approach to treatment because of rapid tumour progression of the tumour. Although there was no evidence of increased MMC-induced chromosome breakage in lymphocytes, stimulation resulted in strongly reduced proliferation. Despite compound heterozygosity, P10 had a severe phenotype (in terms of both dysmorphisms and an early childhood cancer) aligning more closely with the homozygous patients described so far.
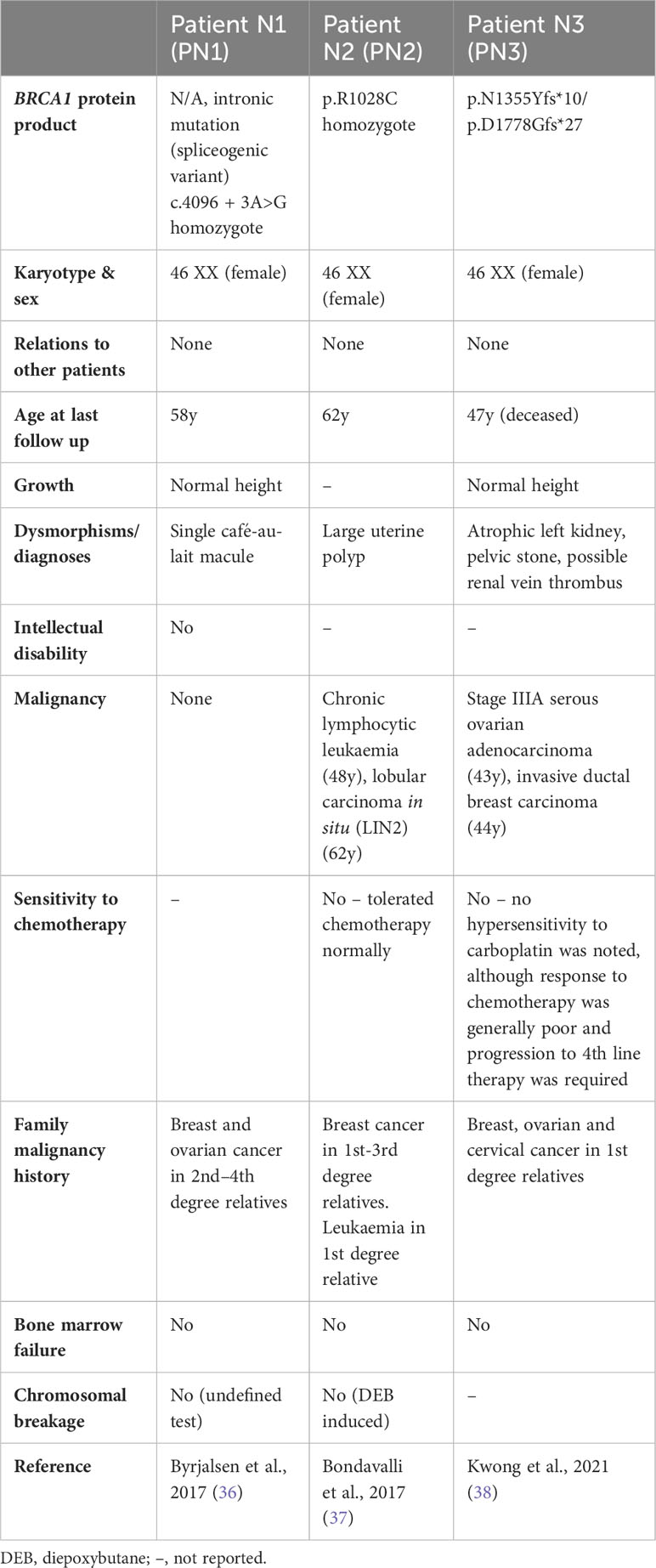
Biallelic BRCA1 mutations without FA-like disorderAt least three individuals (PN1-N3, Table 2) have been reported with biallelic BRCA1 mutations without an FA phenotype. PN1 has a homozygous spliceogenic variant predicted to be highly deleterious, however PN1 has no clinical features consistent with FA or HBOC (BRCA1), suggesting that this variant is likely benign (36). PN2 has a homozygous missense mutation (p.R1028C) predicted to be ‘likely benign’ and has no congenital abnormalities – although chronic lymphocytic leukaemia and breast cancer were reported, suggesting a possible HBOC (BRCA1) phenotype (37). PN3 is a compound heterozygote with breast/ovarian cancers before age 50, but no congenital anomalies – a phenotype more consistent with HBOC (BRCA1) than FA (38). One allele is a known pathogenic mutation (p.N1355Yfs*10), co-occurring with a splice site variant with complex interpretation (p.D1778Gfs*27, Table 3). The sister of PN3 (p.D1778Gfs*27 heterozygote) had breast/ovarian cancers at around 10 years older than the age PN3 developed cancer, possibly suggesting a worsened HBOC (BRCA1) phenotype due to compound heterozygosity.
DiscussionThe emergence of a cohort of patients with pathogenic biallelic BRCA1 mutations and an FA-like phenotype offers insights into the developmental role of BRCA1, as well as broadening the definition of the FA phenotype. There are a number of outstanding questions regarding the underlying biochemistry and pathophysiology of FA-S, as well as a lack of guidance for optimal clinical management. These are likely to be addressed as additional FA-S patients are identified – FA-S may be underreported currently, as FA panels do not typically screen for BRCA1.
It is therefore important that patients with very early breast/ovarian cancer and congenital abnormalities, as well as patients with an FA-like phenotype (particularly in the absence of BMF), are screened for biallelic BRCA1 mutations. Prior to genotyping, many of the FA-S patients described here were investigated for differential diagnoses to explain observed chromosome instability. These include Dubowitz syndrome, non-specific FA, Nijmegen breakage syndrome (NBS), Bloom syndrome and ataxia telangiectasia (AT) (34, 65). NBS, Bloom syndrome and AT are chromosome instability syndromes resulting in cancer predisposition and congenital anomalies, however immunodeficiency is usually present in these cases (unlike in FA) (1, 65). Given the phenotypic variability of FA-S, patients with atypical presentation of these conditions who have not been genetically diagnosed should be considered for BRCA1 testing. Furthermore, the emergence of FA-S as a clinical syndrome may have implications for genetic counselling in families with HBOC (BRCA1). Currently, there is no clear boundary between FA-S and BRCA1-associated hereditary breast and ovarian cancer (HBOC), as exemplified by PN2-3 and P8. These patients are harder to categorise as they have breast and/or ovarian cancer but no evidence of increased chromosomal breakage and minimal dysmorphisms. Presently, these cases are segregated predominantly by their underlying genetics as well as the phenotypic severity (age of cancer, sensitivity to chemotherapeutics and congenital dysmorphisms), however, future study of the functional consequences of these mutations may lead to recategorization. P8 and PN3 are the most borderline cases in terms of both genotype and phenotype and both cases underline that these conditions lie on a continuous spectrum of clinical disease.
One controversy regarding FA-S was the expected lethality of biallelic BRCA1 mutations. The mutations found in this cohort are described in detail in Table 3, including evidence for functional consequences of these mutations, associations with disease in the heterozygous setting (HBOC), and proposed viability mechanisms in these patients. Within the main cohort P1-10, nine out of ten have at least one mutation that could be partially rescued by alternative splicing. One of these (P2) may be rescued by the presence of the naturally occurring Δ9,10 isoform. The remaining eight (P1, P3-7, P9-10) each have at least one mutation that may be rescued by the Δ11q isoform, as proposed by Seo et al., in reference to the homozygous mutations seen in P3-P7 (31, 32). All of the mutations seen in P3-7 lie within, or 3’ of, an alternatively-spliced region, hence allowing unaffected translation of a naturally occurring minor transcript – Δ11q (Figure 2). Δ11q is approximately 40% the length of full-length BRCA1 and consists of normal 5’ and 3’ untranslated regions with truncated exon 11 (c.788_4096del), retaining the reading frame and yielding a shortened, but partially functional, protein isoform (p.263_1365del) (32, 49). The Δ11q isoform has also been shown to underlie a mechanism of resistance to PARP inhibitors and cisplatin in the management of BRCA1 mutated cancers (49). Δ11q isoforms were significantly enriched in fibroblasts from P5 relative to full-length transcripts (compared with control fibroblasts), as a result of nonsense-mediated decay (NMD) of the full-length transcript (32). PN1 and PN2 also have homozygous BRCA1 mutations within the truncated region of exon 11, that could therefore be partially rescued by alternative splicing. However, these may simply be non-pathogenic mutations - the mutation found in PN1 is a spliceogenic VUS with contested pathogenicity, and the mutation in PN2 is not reported to confer significant risk in a heterozygous setting and predicted to be benign. Hence, PN1 and PN2 are not included in the main cohort here – although PN2 could be considered to have an HBOC-like phenotype.
Figure 2 Schematic of full-length (1863 amino acid) BRCA1 protein, with c.787 alternative donor splice site and 11q region (66, 67). Alternative splicing at c.787 resulting in p.263_1365del from protein, Δ11q isoform. Mutations found in FA-S patients shown as red circles, mutations found in patients PN1-3 shown as yellow circles: ◯; missense, ⊘; frameshifts, ⊗; nonsense, ⊕; deletion. Associated proteins shown in blue at approximate points of interaction with BRCA1. RING, zinc finger; NES, nuclear export signal; NLS, nuclear localization sequence; SCD, serine cluster domain; BRCT, BRCA1 C-terminal; P, phosphate.
With regards to compound heterozygous BRCA1 mutations in FA-S patients, most appear to be compatible to life because a hypomorphic missense allele co-occurs with a highly deleterious allele (P1, P2 and P8) hence enough function is retained to enable embryonic survival (29, 30). Previously, it has been considered that the co-occurrence of a VUS in trans with a known pathogenic BRCA1 mutation suggests that the VUS must be benign (68), however the existence of these FA-S patients with viable biallelic mutations challenges this view and has significant implications for how BRCA1 VUS are interpreted. The hypomorphic variants found in trans with highly deleterious mutations in FA-S compound heterozygotes likely confer reduced HBOC risk in a heterozygous setting, as has been suggested for p.V1736A and p.S198Rfs*35 (30) (Table 3). However, alternative splicing may also allow for viability in these cases, as the hypomorphic alleles found in both P1 and P2 can be partially rescued by alternative splicing (Δ11q and Δ9,10 respectively). PN3 is a compound heterozygote with a known deleterious allele in trans with a VUS, whereby the deleterious allele is within the truncated region of exon 11 allowing for a normal Δ11q isoform. PN3 can be confidently considered to have an HBOC-like phenotype, however is not included in the FA-S cohort due to the second mutation being a VUS with unclear pathogenicity, and an overall lower burden of disease compared with P1-10.
P8 is the only case where neither mutation is rescuable by alternative splicing. Despite this, the phenotype of P8 is unexpectedly mild, especially given the known deleterious mutations present in both the RING domain and BRCT domain. It is possible that an alternative rescue mechanism such as interallelic complementation may provide the explanation for this reduced severity phenotype. P8 is tentatively included in the main cohort due to the presence of in trans pathogenic mutations independently associated with HBOC when present in heterozygotes, as well as mild congenital dysmorphisms, sensitivity to chemotherapeutics and having a younger cancer and overall higher severity phenotype compared with PN2-3.
In the case of P9, both variants result in premature termination, however both are 3’ to the alternative donor splice site in exon 11, likely preserving the Δ11q minor transcript. Follow-up with P9 will ascertain whether his phenotype tends towards more severe (as in homozygotes) or less severe (as in most other compound heterozygotes). P10 is an interesting case, as one variant is a truncating mutation 3’ to the splice site (allowing unaffected translation of the Δ11q minor transcript as described above). However, the other variant is downstream of exon 11 (exon 15) so would be included in the Δ11q isoform, likely resulting in NMD of the protein product of this allele. This might explain the severe phenotype seen in P10, which is similar to the most severe homozygotes, as only one allele can produce the partial rescue provided by the Δ11q minor transcript. This is in contrast to homozygotes, where both alleles are able to yield an unaffected Δ11q isoform. Hence, a Δ11q gene dosage effect might underlie the arguably more severe phenotypic presentation in P10 compared to the patients with homozygous mutations.
An interesting observation from the FA-S cases is the presence of breast and ovarian cancers within the cohort. Typically, females with FA do not get breast/ovarian cancers – malignancies are usually haematological (particularly acute myeloid leukaemia and myelodysplastic syndrome) (13), with a smaller proportion of solid (often embryonal) tumours (69), and a high incidence of squamous cell carcinomas in FA patients who reach their third-fourth decade of life (65). There are a number of suggested explanations; females with typical FA tend to be hypogonadal with low serum estrogen and reduced breast/ovarian tissue mass which may be protective against breast/ovarian tumours (70, 71). Additionally, many patients with FA die at a young age, before an age at which breast/ovarian tumours are likely to develop (45% of individuals with FA die from haematological complications before the age of 20) (71, 72). FA-S females may not be hypogonadal, as this was not reported in any of the seven female cases. However, hypogonadism in females is often not reported until teenage years with late puberty, or in milder cases not until presentation of fertility issues.
However, it should be noted that only the female compound heterozygous FA-S patients have presented with breast/ovarian tumours. The female homozygous FA-S patients are all younger, but 3/4 have presented with childhood cancers that align with a more typical FA presentation (astrocytoma, acute lymphoblastic leukaemia and neuroblastoma). P8, the patient with the mildest FA phenotype, is the only FA-S patient reported to have given birth (twin boys). It is unknown whether any fertility treatment was required. All female homozygous FA-S patients are still too young to have been expected to start puberty, and two have died as a result of malignancies. It would be valuable to ascertain whether the surviving female homozygotes (P5 and P7) present with symptoms of hypogonadism. Further to this, given that the homozygous FA-S patients seem to present with a more severe phenotype and ‘FA-like’ cancers, it would be of interest to follow-up with P5 and P7 to investigate whether they are later predisposed to breast/ovarian cancers [as in HBOC (BRCA1)] or not (as in other FA types).
Male hypogonadism does appear to be a feature of FA-S: both males in the cohort presented with cryptorchidism, P9 also with micropenis and low anti-Müllerian hormone. Only 2/10 FA-S patients identified thus far are male (and 0/3 of the non-FA-patients PN1-PN3). With only 10 patients in total this is likely to be coincidental, however possible explanations should be considered in case this observation continues as further FA-S cases are identified. One possibility is that FA-S males with compound heterozygous mutations are at a lower cancer risk than females (as compound heterozygous females seem to develop breast/ovarian cancer) so therefore are not identified. It can be expected that they would also have congenital abnormalities, but in the absence of BMF or malignancy this could easily be misdiagnosed. It is of note that the only male with a homozygous BRCA1 mutation (P6) remains cancer-free despite reaching his late-teens – the oldest homozygous FA-S patient to do so. Another plausible explanation for the relative lack of male FA-S patients could relate to relative viability of male versus female embryos with BRCA1 mutations. Sex ratio distortions (reduced proportion of males) have been reported in the offspring of BRCA1 heterozygous females (73, 74), although the presence/extent of this distortion is disputed (75).
Another striking difference between FA-S patients and classical FA is the absence of BMF, as well as the relative rarity of radial anomalies (only seen in P2 and P6). Although BMF has been considered a hallmark of Fanconi Anaemia, FA-S is not the only type which does not exhibit it - case reports often refer to patients with these presentations as having an ‘FA-like disorder’. Another recently identified rare subtype, FA-O, appears to present similarly to FA-S – including congenital dysmorphisms, sensitivity to crosslinking agents, and without BMF, although only one consanguineous family and one additional individual have been identified (76, 77). In additional, with biallelic FANCM mutations present with an HBOC-phenotype, alongside sensitivity to chemotherapy and possible chromosomal sensitivity but without BMF or congenital abnormalities leading to significant dispute regarding the status of FANCM as a canonical FA protein, as patients (78). Even among more common types of FA, the presentation of BMF is variable – with estimated cumulative incidence of BMF at 10 years ranging from 12.6%-84% depending on predicted risk group (79).
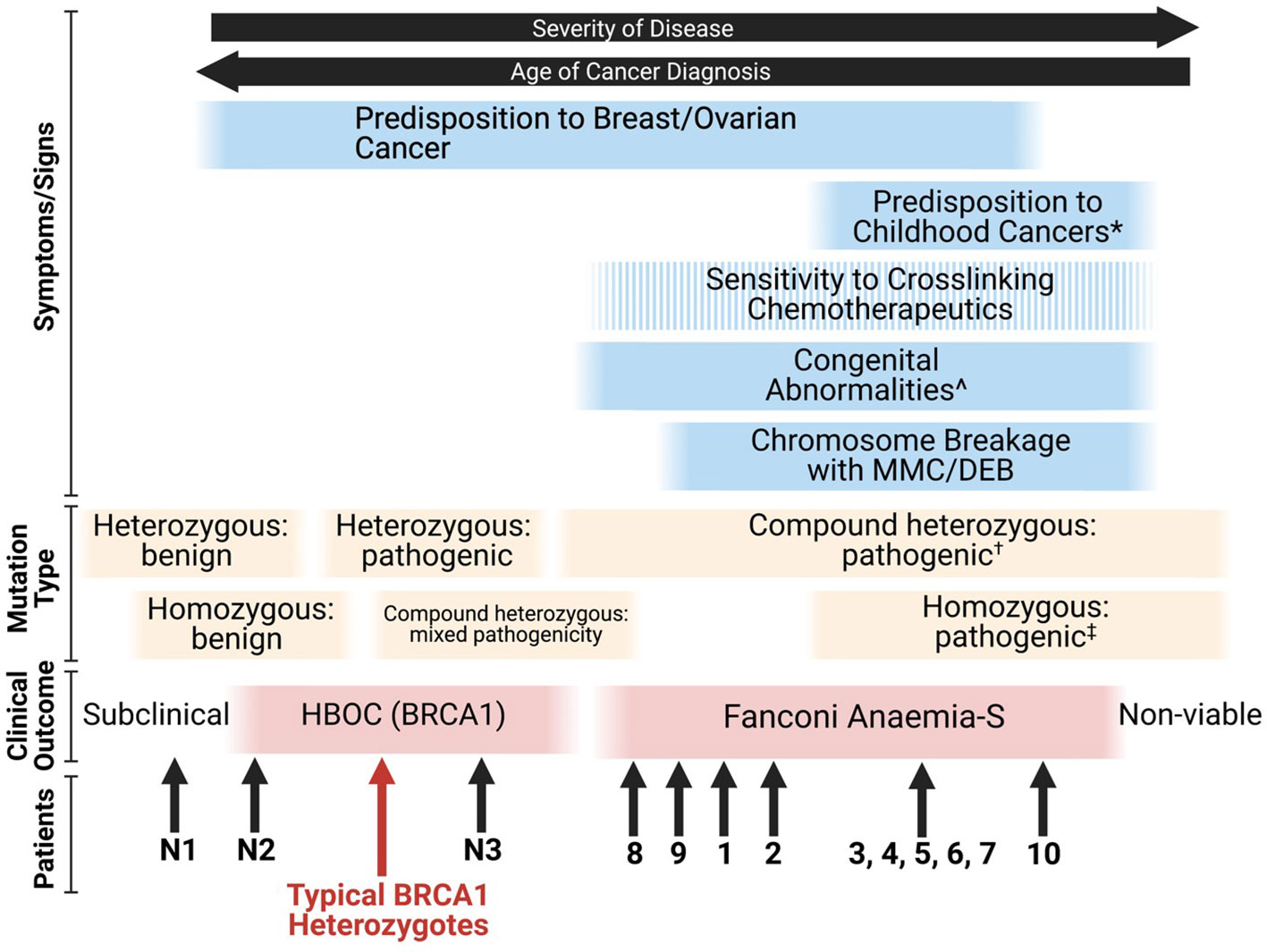
Given that many combinations of deleterious BRCA1 mutations are likely to be embryonically lethal (26), the FA-S phenotype appears to be an intermediate between classical HBOC (BRCA1) and non-viability. The phenotypic spectrum of BRCA1 mutations stretches from subclinical benign monoallelic mutations through to pathogenic homozygous mutations resulting in nonviability of embryos, with some patients blurring the lines between FA-S and HBOC (BRCA1) (Figure 3). From the patient data available, it appears that homozygous mutations tend to result in a more severe FA-like phenotype, whereas compound heterozygosity results in a severe HBOC-like cancer phenotype along with congenital abnormalities. The Δ11q isoform appears to be a key mechanism for survival of biallelic BRCA1 mutations particularly in a homozygous setting, and it is plausible that other splice variants may be subsequently found to provide alternative survival mechanisms.
Figure 3 Proposed phenotypic spectrum of BRCA1 mutations (67). ‘Sensitivity to cross-linking chemotherapeutics’ is a non-ubiquitous presentation among FA-S patients. *; ALL, neuroblastoma and astrocytoma are the three most frequent cancers in children (80). ^; short stature, microcephaly, head/facial dysmorphisms, pigmented skin lesions and intellectual disability. †; two highly deleterious alleles likely result in non-viability, for viability it appears that at least one partially functioning allele is required (or functioning splice variants of at least one allele). ‡; many homozygous pathogenic variants are likely to be non-viable, splice variants may account for viability.
Author contributionsTH: Data curation, Investigation, Methodology, Writing – original draft. AR: Data curation, Formal Analysis, Writing – review and editing.
FundingThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. AR is supported by an NIHR Clinical Lecturership in Paediatric Oncology (CL-2018-13-005).
AcknowledgmentsThe authors wish to thank Dr Lisa Walker for guidance and supervision of TH during her Final Honours Essay, which provided the starting point for this scoping review.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References3. Koc A, JC P, Alikasifoglu M, Joenje H, Altay C. Variable pathogenicity of exon 43del (FAA) in four Fanconi anaemia patients within a consanguineous family. Br J Haematol (1999) 104(1):127–30. doi: 10.1046/j.1365-2141.1999.01156.x
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Chandrasekharappa SC, Lach FP, Kimble DC, Kamat A, Teer JK, Donovan FX, et al. Massively parallel sequencing, aCGH, and RNA-Seq technologies provide a comprehensive molecular diagnosis of Fanconi anemia. Blood (2013) 121(22):e138–48. doi: 10.1182/blood-2012-12-474585
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Joenje H, Levitus M, Waisfisz Q, D'Andrea A, Garcia-Higuera I, Pearson T, et al. Complementation analysis in Fanconi anemia: assignment of the reference FA-H patient to group A. Am J Hum Genet (2000) 67(3):759–62. doi: 10.1086/303067
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, et al. A dominant mutation in human RAD51 reveals its function in DNA interstrand crosslink repair independent of homologous recombination. Mol Cell (2015) 59(3):478–90. doi: 10.1016/j.molcel.2015.07.009
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Ameziane N, May P, Haitjema A, van de Vrugt HJ, van Rossum-Fikkert SE, Ristic D, et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat Commun (2015) 6(1):8829. doi: 10.1038/ncomms9829
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, et al. X-linked inheritance of Fanconi anemia complementation group B. Nat Genet (2004) 36(11):1219–24. doi: 10.1038/ng1458
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Bluteau D, Masliah-Planchon J, Clairmont C, Rousseau A, Ceccaldi R, Dubois d’Enghien C, et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest (2016) 126(9):3580–4. doi: 10.1172/JCI88010
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Mamrak NE, Shimamura A, Howlett NG. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev (2017) 31(3):93–9. doi: 10.1016/j.blre.2016.10.002
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Knies K, Inano S, Ramírez MJ, Ishiai M, Surrallés J, Takata M, et al. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J Clin Invest (2017) 127(8):3013–27. doi: 10.1172/JCI92069
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Garcia-Higuera I, Taniguchi T, Ganesan S, MS M, Timmers C, Hejna J, et al. Interaction of the fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell (2001) 7(2):249–62. doi: 10.1016/S1097-2765(01)00173-3
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Alter BP, Rosenberg PS, Brody LC. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet (2007) 44(1):1–9. doi: 10.1136/jmg.2006.043257
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science (2002) 297(5581):606–9. doi: 10.1126/science.1073834
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Cantor SB, Guillemette S. Hereditary breast cancer and the BRCA1-associated FANCJ/BACH1/BRIP1. Future Oncol (London England) (2011) 7(2):253–61. doi: 10.2217/fon.10.191
CrossRef Full Text | Google Scholar
19. Byrd PJ, Stewart GS, Smith A, Eaton C, Taylor AJ, Guy C, et al. A hypomorphic PALB2 allele gives rise to an unusual form of FA-N associated with lymphoid tumour development. PloS Genet (2016) 12(3):e1005945. doi: 10.1371/journal.pgen.1005945
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Xia B, Dorsman JC, Ameziane N, de Vries Y, Rooimans MA, Sheng Q, et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet (2007) 39(2):159–61. doi: 10.1038/ng1942
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Tischkowitz M, Xia B, Sabbaghian N, Reis-Filho J, Hamel N, Li G, et al. Analysis of PALB2/FANCN-associated breast cancer families. Proc Natl Acad Sci USA (2007) 104(16):6788. doi: 10.1073/pnas.0701724104
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Christou CM, Kyriacou K. BRCA1 and its network of interacting partners. Biol (Basel) (2013) 2(1):40–63. doi: 10.3390/biology2010040
CrossRef Full Text | Google Scholar
24. Liu CY, Flesken-Nikitin A, Li S, Zeng Y, Lee WH. Inactivation of the mouse Brca1 gene leads to failure in the morphogenesis of the egg cylinder in early postimplantation development. Genes Dev (1996) 10(14):1835–43. doi: 10.1101/gad.10.14.1835
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Evers B, Jonkers J. Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene (2006) 25(43):5885–97. doi: 10.1038/sj.onc.1209871
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell (1996) 85(7):1009–23. doi: 10.1016/S0092-8674(00)81302-1
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Gowen LC, Johnson BL, Latour AM, Sulik KK, Koller BH. Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nat Genet (1996) 12(2):191–4. doi: 10.1038/ng0296-191
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Page MJ, Moher D, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. PRISMA 2020 explanation and elaboration: updated guidance and exemplars for reporting systematic reviews. BMJ (2021) 372:n160. doi: 10.1136/bmj.n160
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Domchek SM, Tang J, Stopfer J, Lilli DR, Hamel N, Tischkowitz M, et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov (2013) 3(4):399–405. doi: 10.1158/2159-8290.CD-12-0421
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Sawyer SL, Tian L, Kähkönen M, Schwartzentruber J, Kircher M, University of Washington Centre for Mendelian Genomics, et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov (2015) 5(2):135–42. doi: 10.1158/2159-8290.CD-14-1156
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Freire BL, Homma TK, Funari MFA, Lerario AM, Leal AM, Velloso EDRP, et al. Homozygous loss of function BRCA1 variant causing a Fanconi-anemia-like phenotype, a clinical report and review of previous patients. Eur J Med Genet (2018) 61(3):130–3. doi: 10.1016/j.ejmg.2017.11.003
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Seo A, Steinberg-Shemer O, Unal S, Casadei S, Walsh T, Gumruk F, et al. Mechanism for survival of homozygous nonsense mutations in the tumor suppressor gene BRCA1. Proc Natl Acad Sci USA (2018) 115(20):5241. doi: 10.1073/pnas.1801796115
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Keupp K, Hampp S, Hübbel A, Maringa M, Kostezka S, Rhiem K, et al. Biallelic germline BRCA1 mutations in a patient with early onset breast cancer, mild Fanconi anemia-like phenotype, and no chromosome fragility. Mol Genet Genomic Med (2019) 7(9):e863. doi: 10.1002/mgg3.863
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Chirita-Emandi A, Andreescu N, Popa C, Mihailescu A, Riza AL, Plesea R, et al. Biallelic variants in BRCA1 gene cause a recognisable phenotype within chromosomal instability syndromes reframed as BRCA1 deficiency. J Med Genet (2021) 58(9):648–52. doi: 10.1136/jmedgenet-2020-107198
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Borlin PR, Brazzola P, Frontzek K, Zanoni P, Morscher RJ, Hench J, et al. Cancer in children with biallelic BRCA1 variants and Fanconi anemia-like features: Report of a Malignant brain tumor in a young child. Pediatr Blood Cancer (2022) 69(10):e29680. doi: 10.1002/pbc.29680
留言 (0)