
記住我



The elderly population has risen dramatically which cause the most serious social demographic and medical problem around the world.[1] Advanced age is one of the strongest risk factors for many chronic diseases that negatively impact health and quality of life.[2,3] An increasing body of evidence suggests that age-related changes occur in a regulated manner.[4] Understanding the genetic expression changes that influence aging holds significant importance in mitigating the prevalence of chronic diseases and enhancing the health span of populations.[2,3,5] Therefore, the identification of age-related biomarkers that can predict health status, at an early stage, and screen individuals at high risk of age-related diseases to be targeted for interventions and diagnosis.
Over the last few years, multiple studies have reported that age-dependent changes in gene expression in different tissues may contribute to high susceptibilities to diseases.[4,6–8] Among these tissues, blood offers many advantages as a sample to identify potential biomarker signatures of aging, including convenient acquisition, and reducing the burden of making multiple physiological and clinical assessments.[3,4] Moreover, biomarkers in blood samples may systematically reflect the age-related changes in the internal environment since blood flows through any organ. Thus, we identified blood transcriptomic signatures of aging to evaluate the health status of the elderly in the early stage.
Here, we identified the differentially expressed genes (DEGs) between healthy young and old individuals from human whole blood samples. Molecule functional and pathway analyses were performed to predict the mechanisms that impact the health trajectories. Then, we established a protein-protein interaction (PPI) network to explore potential relationships among the DEGs and screened out key modules and hub genes. Combining the results of the receiver operating characteristic (ROC) curve and the expression analysis of hub genes, we validate the correction between these genes and age. This study allowed us to identify transcriptomic-aging-signature, which will provide new insights into the mechanisms of aging, and provide potential targets for therapies aimed at extending health span.
2. Materials and Methods 2.1. Ethics approvalThis study did not conduct experiments involving animals or humans; therefore, ethical approval was not required.
2.2. Microarray datasets processingThree gene expression profiles (GSE123696, GSE123697, GSE123698) of whole blood in healthy young and old individuals with longitudinal tracking were available from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). These datasets track 135 healthy adults in different age groups with 3 years of follow-up. All data were based on the GPL15207 platform Affymetrix Human Gene Expression Array. The probes were labeled with gene symbols according to the annotation information on GPL15207, and those genes with multiple probes were randomly picked one probe to obtain the gene expression matrix. To enhance the reliability of screening results, the data sets GSE16717 (including 50 old samples and 50 young samples), GSE49058 (including 10 old samples and 10 young samples), and GSE67220 (including 9 old samples and 11 young samples) were added as validation data sets.
2.3. Differential expression selectionWe used the “limma” package in R to screen DEGs between young (< 60 years old) and old (> 60 years old) samples. The threshold values of DEGs were P .value <0.05 and | log2 fold change (FC) | > 0.5. Volcano plots were presented by using the “ggplot” package in R to visualize the results of DEGs. The overlapping DEGs between the 3 profiles were identified by the online visualization software Jvenn (http://jvenn.toulouse.inra.fr/app/example.html).
2.4. Functional and pathway enrichment analysis of DEGsGene ontology (GO) analysis of the overlapping DEGs under the 3 categories, molecular function (MF), cellular component (CC), and biological process (BP) was performed using the “ClusterProfiler” package. A P .value <0.05 was considered statistically significant. Pathway enrichment analysis was performed using the ReactomePA R package.[9] The threshold value for significantly enriched terms was P .value <0.05.
2.5. PPI network of DEGs establishment and hub genes identificationThe protein interactions analysis was performed using the STRING database (http://string-db.org) with confidence > 0.4, and the PPI network was visualized by Cytoscape (v3.9.0). The CytoHubba plug-in in Cytoscape was used to calculate the connectivity of network topology. The key module in the PPI network was identified using the MCODE plugin (degree cutoff = 2, K-core = 2, max depth = 100, and node score cutoff = 0.2), and module genes were identified as hub genes.
2.6. ROC curve analysis of hub genesTo investigate the accuracy of identified hub genes in the GSE123696, GSE123697, and GSE123698 datasets, we performed ROC curve analysis by the “pROC” package. The hub genes, with an area under the curve (AUC) of more than 0.7, were selected to verify their expression. Violin plots were used to visualize the expression profile of hub genes by using the “ggplot2” package.
2.7. Age correlation analysis of hub genesThe correlations of hub genes and ages were verified by using Pearson correlation and the results were shown using Scatterplots with the “ggplot2” package in R.
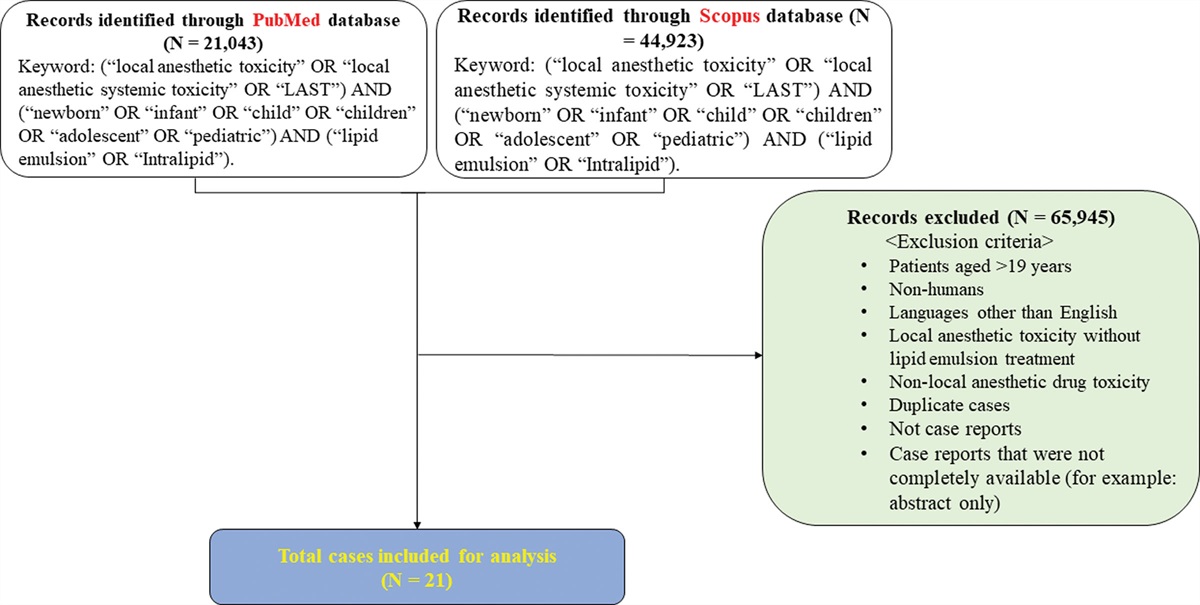
3. Results 3.1. Identification of DEGs between young and old whole blood samplesThree profiles (GSE123696, GSE123697, GSE123698) tracking the gene expression change in healthy young and old whole blood samples during 3 years follow-up were obtained from the GEO database. We screened DEGs with the criteria of P value <0.05 and | log2FC | > 0.5. We obtained 76 DEGs from GSE123696, with 62 downregulated genes and 14 upregulated genes (Fig. 1A). In the gene chip GSE123697, 129 DEGs were obtained, containing 117 downregulated and 12 upregulated genes (Fig. 1B). In addition, 65 DEGs were obtained from GSE123698, 55 of which were downregulated and 10 of which were upregulated (Fig. 1C). The Venn analysis (Fig. 1D) showed that expression changes of 29 genes remain fairly stable during the follow-up period, and thus were chosen as the candidate genes.
 Figure 1.:
Figure 1.: Identification of DEGs. (A–C) Volcano plots of age-related DEGs were identified based on fold-change and P .value of genes with expression differences between young and old samples. The blue dots represent downregulated genes, the red dots represent upregulated genes, and the gray dots represent non-significant genes. (D) An overlap of 29 DEGs showed a similar stable trend in 3 datasets in the Venn diagram. DEGs = differentially expressed genes.
3.2. Functional enrichment analysis of DEGsTo explore the mechanisms and potential roles of the overlapping DEGs, we performed functional and pathway enrichment analyses. GO enrichment analysis was used to enrich DEGs from biological process (BP), molecular function (MF), and CC, respectively. Regarding the BP, DEGs were significantly enriched in negative selection of T cells, humoral immune response, and differentiation of lymphocyte and mononuclear cells (Fig. 2A). Among them, a failure of T cell-negative selection was one of the critical contributors to chronic inflammation during aging.[10] Concerning MF, the most significant terms enriched with DEGs include peptidoglycan binding, immunoglobulin binding, and phosphatidylcholine binding (Fig. 2B). For CC, the GO terms enriched with DEGs were related to age-dependent chronic diseases, including circulating immune complexes, plasma membrane, and blood microparticles (Fig. 2C).[11–13] Moreover, several Reactome pathways related to immune senescence were significantly enriched, including interactions between lymphoid and non-lymphoid cells and their immunoregulatory functions, the Wnt signaling pathway, and FCGR activation[14–17] (Fig. 2D), suggesting that immunosenescence plays a pivotal role in threatening the health of the elderly.
 Figure 2.:
Figure 2.: Enrichment analysis of DEGs. (A–C) The bar plots show the DEGs enriched in the top 10 GO categories of BPs, MFs, and CCs, respectively. The x-axis stands for a number of enriched genes. (D) The top 10 Reactome pathways enrichment of DEGs. BP = biological process. BP = biological process, CC = cellular component, DEGs = differentially expressed genes, MF = molecular function.
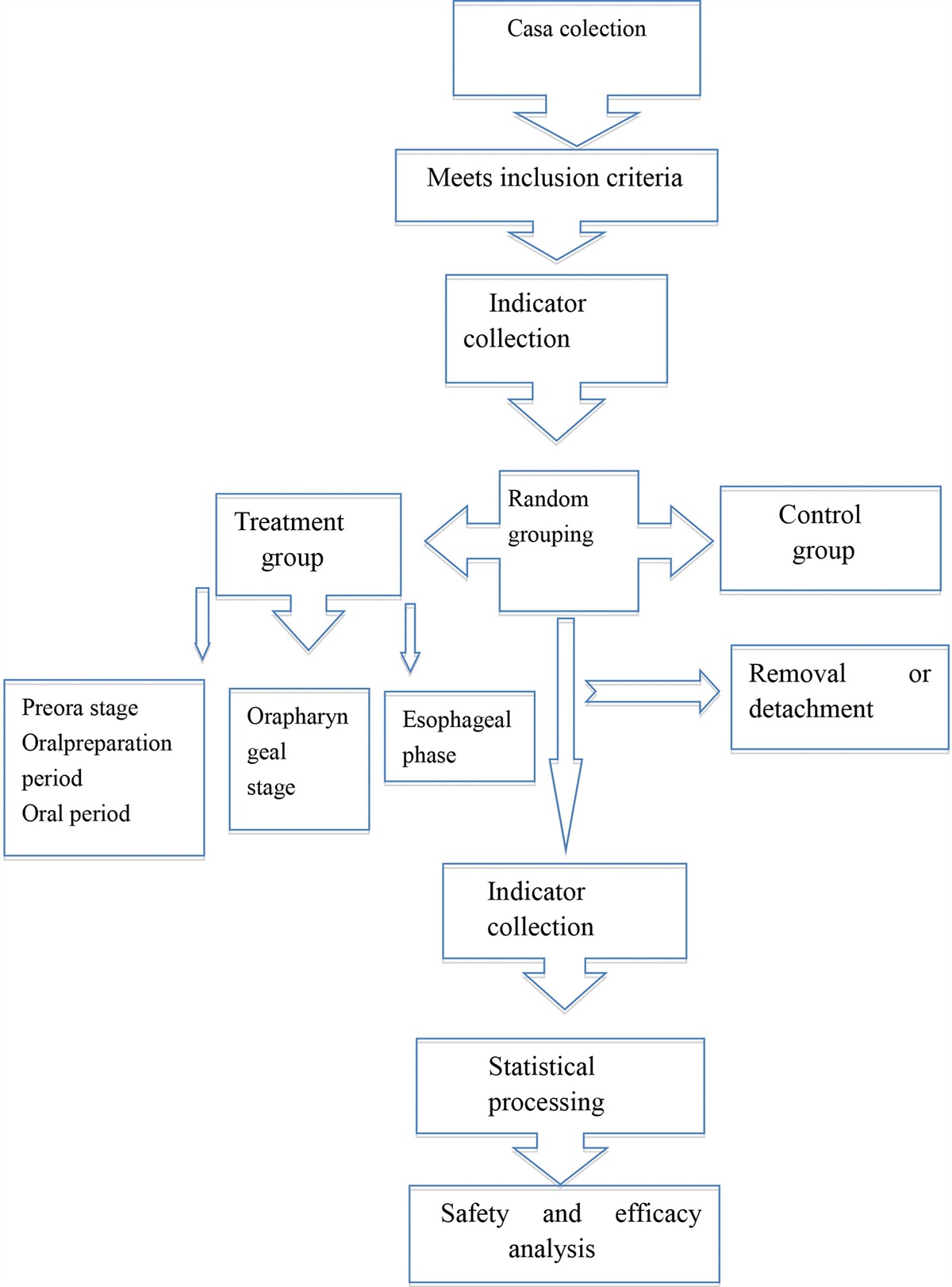
3.3. PPI network establishment and hub genes identificationTo explore the interactive relationships involved in DEGs, a PPI network with 13 nodes and 16 edges was constructed (Fig. 3A). The module analysis of the network was performed using the “MCODE” plug-in of Cytoscape, only one module with 7 genes and 10 edges was obtained (Fig. 3B). Then we used 3 topological methods of the CytoHubba program in Cytoscape to screen hub genes. There are 4 genes in all 3 methods, including Bach2, IGLL5, Jchain, and POU2AF1 (Fig. 3C), suggesting they may play vital roles in the aging process. Therefore, these 4 hub genes were selected for further research.
 Figure 3.:
Figure 3.: The 4 hub genes screened from the PPI network. (A) The PPI network of DEGs was constructed by using The STRING database. (B) The node gene cluster was identified by the MCODE plug-in in Cytoscape. (C) The key modules genes (hub genes) were screened by 3 algorithms Degree, MCC, and MNC.
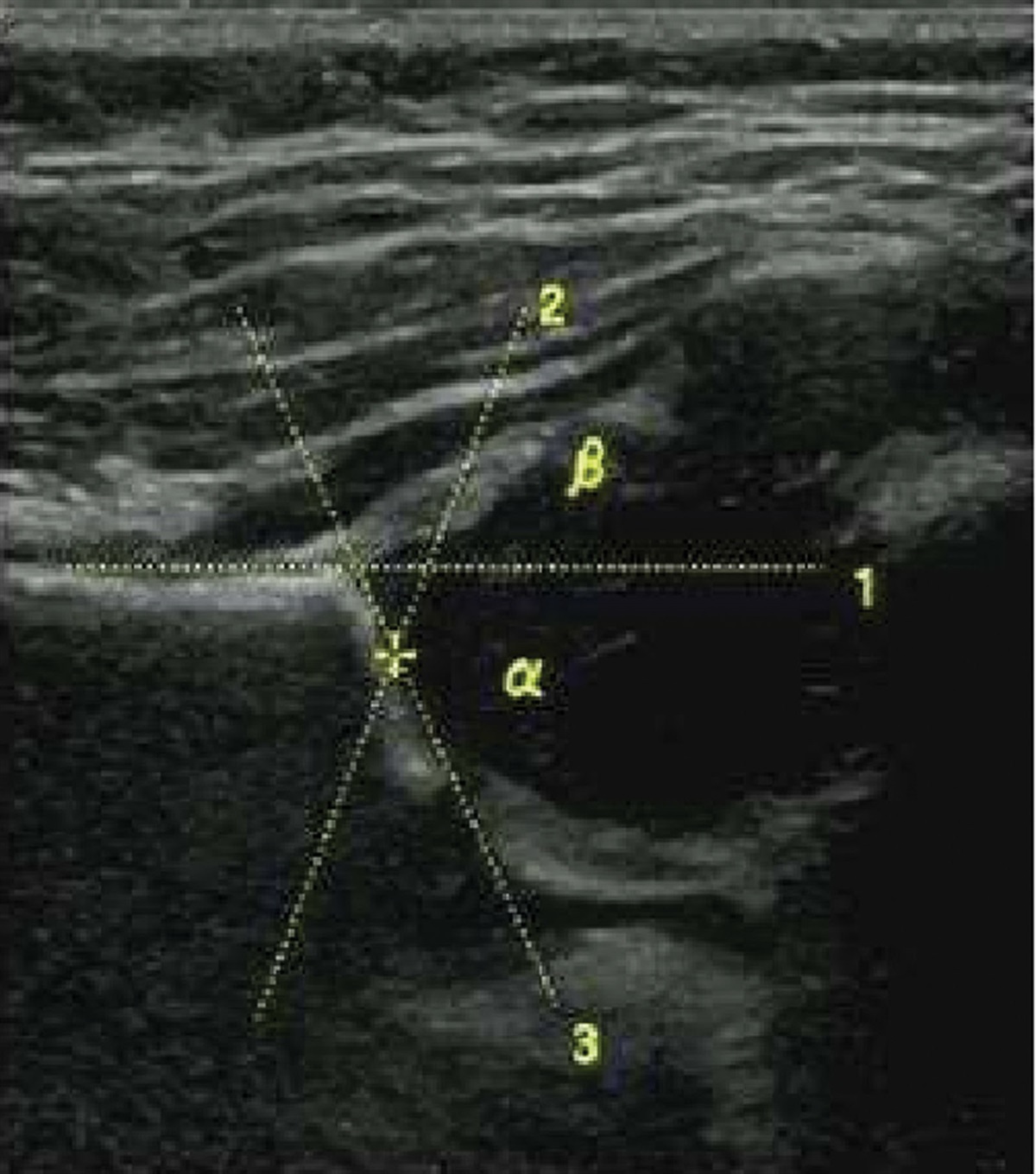
3.4. Correlation analysis between expression of hub genes and chronological ageIn order to better understand the role of 4 hub genes in the process of aging, we performed a Pearson analysis between hub gene expression and age. Correlation analysis showed that the mRNA expression levels of IGLL5, POU2AF1, Jchain, and Bach2 in human blood samples were negatively correlated with chronological age (Fig. 4), suggesting that these genes may restrain the procession of aging.
 Figure 4.:
Figure 4.: Explorations between chronological age and hub genes. (A) The correlation between mRNA expression of IGLL5 and age in 3 datasets. (B) The correlation between mRNA expression of POU2AF1 and age in 3 datasets. (C) The correlation between mRNA expression of Jchain and age in 3 datasets. (D) The correlation between mRNA expression of Bach2 and age in 3 datasets.
3.5. Analysis of expression and ROC curve of hub genesThe violin plots were used to visualize the expression of IGLL5, POU2AF1, Jchain, and Bach2 in the 3 datasets. As shown in Figure 5 and Supplementary Figure 1 (https://links.lww.com/MD/K939), the expression levels of 4 hub genes were significantly decreased in elderly individuals compared to the young group. To further evaluate the diagnostic performance of these hub genes in the degree of aging, ROC curves of GSE123696, GSE123697, and GSE123698 were performed. The AUC values of 3 hub genes (IGLL5, POU2AF1, and Bach2) were steadily higher than 0.7 in 3 datasets, while Jchian was lower than 0.7 in GSE123698 (Fig. 6). These results indicated that the genes IGLL5, POU2AF1, and Bach2 possessed more significant assessment values in aging than Jchian.
 Figure 5.:
Figure 5.: The expression level of 4 hub genes. (A) IGLL5 expression levels in old and young groups in 3 datasets. (B) POU2AF1expression levels in old and young groups in 3 datasets. (C) Jchain expression levels in old and young groups in 3 datasets. (D) Bach2 expression levels in old and young groups in 3 datasets.
 Figure 6.:
Figure 6.: Age-specific effect of 4 hub genes. (A) ROC curve analysis of IGLL5 in 3 datasets. (B) ROC curve analysis of POU2AF1 in 3 datasets. (C) ROC curve analysis of Jchain in 3 datasets. (D) ROC curve analysis of Bach2 in 3 datasets. AUC = the area under the ROC curve, ROC = receiver operating characteristic.
4. DiscussionAging is the greatest risk factor for the prognosis of most chronic diseases that decrease patients’ quality of life, including cancer, depressive disorder, and respiratory diseases. Increasing evidence suggests that aging affects dynamic changes in gene expression which interferes regulation of cell-signaling pathways and can increase diseases.[4,18,19] Therefore, the identification of age-associated gene expression signatures by transcriptomic analysis was conducted to identify individuals whose health deteriorates rapidly with aging.
In this study, we used transcriptomic analysis to identify 29 overlapping DEGs between 3 profiles according to age. We performed functional and biological pathway enrichment analysis of these DEGs to explore the mechanisms that impact the progression of aging. We found that these genes were mainly enriched in the regulation of immune response and activation of inflammatory pathways, which not only lead to poor health but also can threaten the quality of life in older persons. To explore the potential relationships among DEGs and identify age-related biomarkers, we performed a PPI network and acquired 4 hub genes (IGLL5, Jchain, POU2AF1, and Bach2). Combined with their high correlations of chronological age, we speculated that IGLL5, Jchain, and POU2AF1 might be the key genes to accelerate the procession of aging. Moreover, ROC curves of hub genes showed that down-regulated expressions of IGLL5, POU2AF1, and Bach2 possess better assessment values in the procession of aging than Jchain. These results were accomplished to offer new insights into understanding the mechanisms of aging from the transcriptomic level and potential targets for anti-aging intervention.
IGLL5, encoding one of the immunoglobulin lambda-like polypeptides, frequently occurs in the mutation in cancer, including lymphoma, multiple myeloma, and chronic lymphoblastic leukemia.[20–22] Although the mutation rate of IGLL5 did not change with age,[23] Yao et al have shown that the low expression level of IGLL5 is a good diagnostic in elderly patients with late-onset major depressive disorder which increases the risk of suicide and disability in olds.[24] Therefore, we speculated that IGLL5 has a vital role in staying healthy for elders. Our study found that IGLL5 showed a down-regulated level in elders with a great ROC score (AUC value > 0.7), further illustrating the potential of IGLL5 as a biomarker of health for elders.
POU2AF1 (POU domain class 2-associating factor 1), a transcriptional coactivator, plays a vital role in B-cell responses to antigens and regulates other host defense-related genes.[25] The expression of POU2AF1 is associated with the severity of emphysema in chronic obstructive pulmonary disease patients which is one of the serious respiratory diseases that threaten the health of the elderly, whose prevalence increases with age.[26,27] In our study, we found that the expression level of POU2AF1 was negatively correlated with age, suggesting that POU2AF1 was a critical contributor to weakened respiratory defenses in the old group.
The transcription factor Bach2, which belongs to the BTB and CNC homology (Bach) family, is reported to regulate aging in various ways.[28] As a highly sensitive DNA damage responder, the expression level of Bach2 was down-regulated during aging.[29] Moreover, Bach2 has been reported to affect immunosenescence since its expression was negatively associated with age in human immune cells.[30] Our study also suggested that Bach2 mRNA was decreased in elders with the highest AUC value, which supports the previous results.
Immunoglobulin J polypeptide (Jchain/IGJ), as a plasma cell-restricted gene, has been recognized as an important prognostic marker of immune-mediated diseases.[31,32] Recently, studies reported that the gene expression trajectories of Jchain were highly correlated with deleterious processes of senescence. The expression of Jchain increased steadily in organs responsible for generating adaptive immune cells, such as the spleen, bone, and marrow, this may be associated with the origin of the age-related inflammation.[33] Instead, we found that Jchain mRNA in blood decreased with the age of the healthy persons, this reflected a regression of the body immunity with age may be caused by adaptive immune cell loss. The result further indicated that appropriately improving levels of adaptive immune cells may effectively delay aging effects.
Altogether, we identified 29 age-associated mRNAs that might create a causal environment for age-associated diseases. Functional and pathway analyses of these genes may further shed light on the mechanisms of aging. Finally, we screened 4 key genes (IGLL5, Jchain, POU2AF1, and Bach2) which were more likely to maintain a healthy body in older age. In addition, these hub genes not only were new aging biomarker candidates but also highlighted important mechanisms of aging and provided potential targets for pharmacological intervention.
Nevertheless, this study has several limitations. At present, our findings were based on publicly microarray data and verified the expression levels of the identified hub genes in independent datasets, without experimental or clinical confirmation. In the future, experiments will be needed to explore the roles and molecular mechanisms of these genes in the aging process. Moreover, the profiles we used only tracked gene expression in a fixed population which may cause bias in the results. Therefore, multiple datasets will be needed to improve the integrity and reliability of the results. Notwithstanding these limitations, the results of this study provide some new insights into the mechanisms and potential interventional targets of aging.
AcknowledgmentsWe appreciate all investigators and staff in this study.
Author contributionsConceptualization: Yan Zhang.
Formal analysis: Yan Zhang.
Investigation: Yan Zhang, Chonghui Liu.
Methodology: Yan Zhang, Chonghui Liu.
Supervision: Chonghui Liu.
Visualization: Yan Zhang.
Writing – original draft: Yan Zhang.
Writing – review & editing: Yan Zhang, Chonghui Liu.
References [1]. Rudnicka E, Napierala P, Podfigurna A, et al. The World Health Organization (WHO) approach to healthy ageing. Maturitas. 2020;139:6–11. [2]. Tanaka T, Basisty N, Fantoni G, et al. Plasma proteomic biomarker signature of age predicts health and life span. eLife. 2020;9:e61073. [3]. Johnson LC, Parker K, Aguirre BF, et al. The plasma metabolome as a predictor of biological aging in humans. GeroScience. 2019;41:895–906. [4]. Shavlakadze T, Morris M, Fang J, et al. Age-related gene expression signature in rats demonstrate early, late, and linear transcriptional changes from multiple tissues. Cell Rep. 2019;28:3263–3273.e3. [5]. Lowsky DJ, Olshansky SJ, Bhattacharya J, et al. Heterogeneity in healthy aging. J Gerontol A Biol Sci Med Sci. 2014;69:640–9. [6]. Pagiatakis C, Musolino E, Gornati R, et al. Epigenetics of aging and disease: a brief overview. Aging Clin Exp Res. 2019;33:737–45. [7]. Yu Y, Fuscoe JC, Zhao C, et al. A rat RNA-Seq transcriptomic BodyMap across 11 organs and 4 developmental stages. Nat Commun. 2014;5:3230. [8]. Yue T, Chen S, Zhu J, et al. The aging-related risk signature in colorectal cancer. Aging (Albany NY). 2021;13:7330–49. [9]. Yu G, He Q-Y. ReactomePA: an R/bioconductor package for reactome pathway analysis and visualization. Mol Biosyst. 2016;12:477–9. [10]. Coder BD, Wang H, Ruan L, et al. Thymic involution perturbs negative selection leading to autoreactive T cells that induce chronic inflammation. J Immunol. 2015;194:5825–37. [11]. Nicolson GL, Ferreira de Mattos G. A brief introduction to some aspects of the fluid-mosaic model of cell membrane structure and its importance in membrane lipid replacement. Membranes (Basel). 2021;11:947. [12]. Kim Y, Goodman MD, Jung AD, et al. Microparticles from aged packed red blood cell units stimulate pulmonary microthrombus formation via P-selectin. Thromb Res. 2020;185:160–6. [13]. Arinola OG, Rahamon S, Edem VF, et al. Elevated plasma level of circulating immune complexes in Nigerian adults infected with severe acute respiratory syndrome corona virus-2. Arch Basic Appl Med. 2021;9:69–72. [14]. Marrella V, Facoetti A, Cassani B. Cellular senescence in immunity against infections. Int J Mol Sci . 2022;23:11845. [15]. Gasparoto TH, Dalboni TM, Amor NG, et al. Fcgamma receptors on aging neutrophils. J Appl Oral Sci. 2021;29:e20200770. [16]. Kim C, Jin J, Weyand CM, et al. The transcription factor TCF1 in T cell differentiation and aging. Int J Mol Sci . 2020;21:6497. [17]. Budamagunta V, Foster TC, Zhou D. Cellular senescence in lymphoid organs and immunosenescence. Aging (Albany NY). 2021;13:19920–41. [18]. Lin H, Lunetta KL, Zhao Q, et al. Whole blood gene expression associated with clinical biological age. J Gerontol A Biol Sci Med Sci. 2019;74:81–8. [19]. Jeong I, Lim J-H, Park J-S, et al. Aging-related changes in the gene expression profile of human lungs. Aging (Albany NY). 2020;12:21391–403. [20]. Li SS, Zhai XH, Liu HL, et al. Whole-exome sequencing analysis identifies distinct mutational profile and novel prognostic biomarkers in primary gastrointestinal diffuse large B-cell lymphoma. Exp Hematol Oncol. 2022;11:71. [21]. White BS, Lanc I, O’Neal J, et al. A multiple myeloma-specific capture sequencing platform discovers novel translocations and frequent, risk-associated point mutations in IGLL5. Blood Cancer J. 2018;8:35. [22]. Kasar S, Kim J, Improgo R, et al. Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat Commun. 2015;6:8866. [23]. Zhao A, Wu F, Wang Y, et al. Analysis of genetic alterations in ocular adnexal mucosa-associated lymphoid tissue lymphoma with whole-exome sequencing. Front Oncol. 2022;12:817635. [24]. Yao PA, Sun HJ, Li XY. Identification of key genes in late-onset major depressive disorder through a co-expression network module. Front Genet. 2022;13:1048761. [25]. Zhou H, Brekman A, Zuo WL, et al. POU2AF1 functions in the human airway epithelium to regulate expression of host defense genes. J Immunol. 2016;196:3159–67. [26]. Faner R, Cruz T, Casserras T, et al. Network analysis of lung transcriptomics reveals a distinct B-cell signature in emphysema. Am J Respir Crit Care Med. 2016;193:1242–53. [27]. Zhong S, Yang L, Liu N, et al. Identification and validation of aging-related genes in COPD based on bioinformatics analysis. Aging (Albany NY). 2022;14:4336–56. [28]. Zhou Y, Wu H, Zhao M, et al. The bach family of transcription factors: a comprehensive review. Clin Rev Allergy Immunol. 2016;50:345–56. [29]. Uittenboogaard LM, Payan-Gomez C, Pothof J, et al. BACH2: a marker of DNA damage and ageing. DNA Repair (Amst). 2013;12:982–92. [30]. Chi VLD, Garaud S, De Silva P, et al. Age-related changes in the BACH2 and PRDM1 genes in lymphocytes from healthy donors and chronic lymphocytic leukemia patients. BMC Cancer. 2019;19:81. [31]. Cruz-Rodriguez N, Combita AL, Enciso LJ, et al. Prognostic stratification improvement by integrating ID1/ID3/IGJ gene expression signature and immunophenotypic profile in adult patients with B-ALL. J Exp Clin Cancer Res. 2017;36:37. [32]. Gui S, O’Neill WQ, Teknos TN, et al. Plasma cell marker, immunoglobulin J polypeptide, predicts early disease-specific mortality in HPV+ HNSCC. J ImmunoTher Cancer. 2021;9:e001259. [33]. Schaum N, Lehallier B, Hahn O, et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature. 2020;583:596–602.
留言 (0)