Cell lines
Multiple myeloma cell lines, MM.1S and RPMI 8226, were obtained from the Cell Bank of Chinese Academy of Sciences. MM.1S-luc cells were MM.1S cells lentivirally transduced with firefly luciferase and GFP, and sorted for GFP positivity for cytotoxicity assays. The same protocol was used to generate RPMI 8226-luc and CHO-luc cells. All MM cell lines were cultured in RPMI 1640 media and 10% fetal bovine serum (FBS). LentiX-293T cells for lentiviral production were grown in Dulbecco’s Modified Eagle Medium (DMEM) medium containing 10% FBS. CHO cells were using serum-free chemically defined (CD) media (Thermo Fisher 10743029). All cell lines were cultured at 37 °C, 5% CO2.
Human samples
The healthy donor peripheral blood mononuclear cells (PBMCs) were obtained from HemaCare Corporation or AllCells.
Identification and engineering of the nanobody tandem
We immunized two llamas with purified BCMA, yielding a high-volume nanobody phage display library from which we isolated and sequenced hundreds of anti-BCMA nanobody candidates. We ranked 41 clones based on their affinities to the extracellular domain of BCMA and constructed monovalent CAR vectors with individual VHHs. Despite the simplicity of the 54-residue BCMA ectodomain, we identified 8 VHH clones with differential epitope binding capabilities, leading to the construction of 38 multi-epitope CAR T constructs. After further cytotoxicity assessments, the Nb1-Nb2 tandem was chosen for cilta-cel due to their highest overall affinity and differential individual binding epitopes.
Protein expression and purification
Purified Nb1, Nb2, and NbT were provided by GenScript (Nanjing, China). Briefly, the corresponding coding sequence of Nb1, Nb2, or NbT was each cloned into the pET-22b vector in frame with a carboxyl-terminal 6× His tag. These constructs were transformed into BL21 E. coli cells. The cells were cultivated in Luria-Bertani (LB) broth supplemented with ampicillin (100 μg/ml final concentration). The expression of nanobodies was induced with 500 µM (final concentration) isopropyl β-D-1-thiogalactopyranoside (IPTG) at an optical density of around 1.2, and the cultures were grown at 15 °C overnight. Cells were harvested and resuspended in a lysis buffer containing 20 mM Tris-HCl (pH 8.0) and 100 mM NaCl. After osmotic shock and centrifugation, the nanobodies were purified from the supernatant by nickel affinity chromatography and polished using a Superdex 75 column (Cytiva) in 20 mM Tris-HCl (pH 8.0) and 100 mM NaCl. BCMAECD (residues 1–54 of human BCMA; UniProt accession code: Q02223-1) was synthesized by GL Biochem (Shanghai).
Surface plasmon resonance (SPR)
Surface plasmon resonance (SPR) binding experiments were performed using a Biacore 8 K system (Cytiva) at 25 °C in 10 mM HEPES (pH 7.4), 150 mM NaCl, and 0.05% (v/v) Tween 20. Fc-tagged and His-tagged human BCMAECD immobilized via amine coupling onto CM5 sensor chips as ligands. Five concentrations of Nb1, Nb2, or NbT obtained by two-fold gradient dilution were flowed over the chip surface as the analytes. To test whether the two proteins compete for BCMA binding, Fc-tagged human BCMAECD was captured onto a Protein A sensor chip surface via the Fc tag. To analyze the direct competitive binding characteristics of Nb1 and Nb2, Nb1/Nb2 protein was loaded followed by the mixture of Nb1 and Nb2. To analyse the binding characteristics of Nbs and BCMA ligands, Nb1/Nb2/NbT were first saturated until a binding steady state was reached. Afterward, the human BCMA ligands APRIL or BAFF were injected in the presence of Nb1/Nb2/NbT. All analyses were carried out in Biacore Insight Evaluation software (Cytiva).
Sedimentation-velocity analytical ultracentrifugation
Sedimentation velocity was performed with an XL-I analytical ultracentrifuge (Beckman Coulter) equipped with a four-cell An-60 Ti rotor for molecular weight analysis of BCMAECD, NbT, and NbT–BCMAECD complex. For other samples, including Nb1, Nb2, Nb1–BCMAECD complex, and Nb2–BCMAECD complex, an eight-cell An-50 Ti rotor was used. Protein complexes were generated by mixing Nb1, Nb2, or NbT with BCMAECD respectively in a molar ratio of 1:4, followed by incubation overnight at 4 °C. All samples were prepared in 1 mg/ml 400 μl for analysis and applied at a speed of 45,000 rpm in 20 mM Tris pH 8.0, 100 mM NaCl, at 4 °C. Absorbance scans were taken at 280 nm at the intervals of 0.003 cm size in a radial direction. The different c (s) and theoretical molecular weights were calculated by SEDFIT software.41
Crystallization
The purified Nb1 or Nb2 and synthesized human BCMAECD were mixed at a molar ratio of 1:4. The mixture was incubated at 4 °C overnight and further purified using a Superdex 75 column (Cytiva). Initial crystallization trials were performed by the sitting-drop vapor diffusion method using a Mosquito (Art Robbins) crystallization robot at 20 °C. The protein solution, with a concentration of 10 mg/mL, and the reservoir solution were mixed in a 1:1 (v/v) ratio. Nb1–BCMAECD complex crystals for data collection were grown from 0.2 M ammonium sulfate, 20% w/v polyethylene glycol 3350, pH 6.0. The best crystals of the Nb2-BCMAECD complexes were grown with a well buffer containing 1.8 M sodium acetate trihydrate pH 7.0 and 0.1 M Bis-Tris propane pH 7.0. Crystals reached full size within three days and were harvested using 20% (v/v) glycerol as cryo-protectant, flash-frozen, and stored in liquid nitrogen for data collection.
Data collection and structure determination
X-ray data were collected on beamlines BL17U1 and BL19U1 at Shanghai Synchrotron Radiation Facility (SSRF) at 100 K and at a wavelength of 0.97853 Å using a Pilatus3 6 M image plate detector. Data integration and scaling were performed using the program XDS.42 The structure of the Nb1-BCMA or Nb2-BCMA complex was determined by molecular replacement using the previously reported structures (PDB: 5BOP and 1XU2) as search models using the program PHASER.43 The output models from molecular replacement were subsequently subjected to iterative cycles of manual model adjustment with Coot44 and refinement was finished with Phenix.45 Data collection and structure refinement statistics are summarized in table S1.
Small-angle X-ray scattering (SAXS)
Nb1–BCMAECD and Nb2–BCMAECD complex were prepared as described in crystallization. The NbT and BCMAECD were mixed at a molar ratio of 1:4. After incubation at 4 °C overnight, the mixtures were further purified by Superdex 200 column (Cytiva) gel filtration in 20 mM Tris pH 8.0, 100 mM NaCl buffer. SAXS experiments were performed at beamline BL19U2 of the National Facility for Protein Science Shanghai at SSRF. The wavelength, λ, of X-ray radiation, was set as 1.033 Å. Three concentrations were measured and SAXS data were collected at 25 °C using 60 μL sample as 20 × 1 s exposures. Data analysis was performed using BioXTAS RAW46 and ATSAS software package.47
MM1.S proliferation assay
For proliferation assay, MM.1S cells were serum starved in RPMI 1640 media overnight. Then MM.1S cells were cultured for 3 days in RPMI 1640 media containing 2% FBS with APRIL (400 ng/mL) or BAFF (400 ng/mL) in the presence or absence of Nb1 (10 μg/mL), Nb2 (10 μg/mL), and NbT (10 μg/mL). The cells were detected by CCK-8 Assay Kit (Vazyme, A311-02) according to the protocol.
Lentivirus package and titer
LentiX-293T cells (Clontech) were used for lentivirus production. 2 × 107 LentiX-293T cells were seeded into each 15 cm dish before transfection. The next day, adherent cells with 80% confluence were accepted for transfection to obtain optimal lentivirus packaging efficiency. The transfection plasmid cocktails which included pMD2.G, pMDLg/pRRE, pRSV-Rev, and each transfer plasmid plasmids (pCDH-Nb1CAR, pCDH-Nb2CAR, pCDH-NbTCAR) were mixed by gently pipetting. PEI reagents were added to the mixture at a volume ratio of 3:1. 48 h post-transfection, the supernatant was collected and concentrated by ultracentrifugation to obtain the lentivirus. 5 × 106 CHO cells in 2 mL were added to 6 well plates and serially diluted lentivirus was added into each well to initiate the transduction. 3 days later, the cells of each well were collected and stained with BCMA-FITC (ACRO, BCA-HF254) for 45 min followed by a flow cytometry assay to evaluate the virus infection titer.
CAR T cell production
Human T cells were isolated from healthy donor PBMCs using Pan T Cell Isolation Kit (Miltenyi Biotec, 130-096-535). The purified T cells were stimulated for 48 h using T Cell TransAct reagents (Miltenyi Biotec 130-111-160) following the manual instruction. Lentivirus was mixed with T cell suspension at 5 MOI (multiplicity of infection). The bulk T cells were cultured using TexMACS GMP medium supplemented with IL-2. After seven days, CAR T cells were harvested and the transduction efficiency was assessed by fluorescence-activated cell sorter (FACS) of cells stained for BCMA-FITC (ACRO, BCA-HF254). We generated different CAR T cells, including Nb1 CAR T, Nb2 CAR T, NbT CAR T, and untreated (UT) cells, using T cells from the same healthy donor on one experimental setting.
CAR T cell in vitro cytotoxicity co-culture assay
In cytotoxicity assays, CAR T cells were co-incubated with target cell (MM.1S-luc cells, RPMI 8226-luc cells) at an effector-to-target (E: T) ratio of 1:1 or 4:1 for 24 h. The number of CAR T cell was consistent in each group and was complemented with UT cells from the same donor according to the CAR positivity. Controls were UT cells from the same donor. After 24 h incubation in 37 °C, 5% CO2 cell culture incubator, 50 μL supernatants were collected for further cytokine release assay. And the cells were added with 100 μl ONE-Glo Firefly luciferase assay reagent mix (Promega, E6120) and incubated at room temperature for 1 min. The remaining living target cells were counted as a relative light unit (RLU) in a microplate reader (Tecan Spark 10 M). The cytotoxicity of CAR T on target cells was calculated with the formula as \(}=\frac}_}-}_}}}_}-}_}}\). The supernatant was collected for cytokines detection using LEGENDplex™ kits Human CD8/NK Panel kit according to the manufacturer’s protocol (BioLegend). Data were collected by using the BD LSRFortessa and analyzed with LEGENDplex v8.0. For low BCMA levels cytotoxicity assay, CAR T cells or UT cells were cocultured at the effector (CAR T or UT) to target cell (CHO-BCMA-Luc) ratio of 4:1.
Imaging flow cytometry
The analysis of immune synapse formation between CAR T cells and MM.1S used imaging flow cytometry on Amnisr® ImageStream®X MK II. Different CAR T cells were co-cultured with MM.1S for 15 min at 37 °C with a 1:1 effector-to-target ratio. After washing cells with PBS, cells were stained with antibodies against CD3-APC-A750 (Beckman A94680), CD138 APC (Beckman A87787) for 30 min at 4 °C. After fixation and permeating, cells were stained with Phalloidin Alexa Fluor 488 (Thermo Fisher A12379) and DAPI (Santa cruz, sc-3598) at 4 °C. Data analysis was performed using Amnis IDEAS software (version 6.2). The analysis strategy we used was described previously.48,49 Briefly, MM.1S cells were gated first based on CD138 positive fluorescence intensity, and then conjugates of CD3, CD138 double positive cells were identified. Conjugates were used to identify adherent cells according to a strategy of Area_M01 larger than 250 units and aspect ratio lower than 0.82. For counting of immune synapse, CD3 T cells were selected to create a mask of CD3 image (“T-cell mask” = Threshold (M12, Ch12, 60)), and DAPI was selected for “valley mask” (valley (M07, Ch07,3)), and then “T-cell synapse mask” was defined (“T-cell mask” AND “valley mask”). Immune synapse formation was determined by an over 30% enrichment of F-actin in the “T-cell synapse mask”.
Generation of CHO-BCMA-luc cells
BCMA coding sequence were retrieved from NCBI database (access number NM_001192.3) and cloned to the pUC57 vector GenScript (Nanjing, China). RNA (IVT-RNA) was prepared in vitro using the mMESSAGE mMACHINE™ T7 Transcription Kit (Invitrogen AM1344), and added poly(A) tail to RNA transcripts using Poly(A) Tailing Kit (Invitrogen, AM1350) to enhance translation initiation efficiency, then purified by the RNeasy Mini Kit (Qiagen, 74104) according to the manufacturer’s protocol. To generate CHO-BCMA-luc cells with escalating cell-surface BCMA levels, different amounts (0 μg, 0.3125 μg, 1.25 μg, or 5 μg) of prepared BCMA RNA were delivered into CHO-luc cells using electroporation: for each sample, 5 × 106 CHO-luc cells were harvested and washed with phosphate-buffered saline once, resuspended in 120 μl electroporation buffer contained in an electroporation kit (Celetrix 1204), then transfected by electroporation at 700 V for 30 ms using a cell electroporator (Celetrix CTX-1500A).
Quantitative analysis of cell surface BCMA antigen expression
Gradational BCMA expressed CHO-BCMA-luc cells, RPMI 8226-luc, MM.1S-luc, and five beads population of Quantum™ Simply Cellular® (Bio-rad, Cat No.815) were stained with a monoclonal anti-human CD269 (BCMA) antibody conjugated with PE (phycoerythrin) (BioLegend 357504). CHO-BCMA-luc, RPMI 8226-luc, and MM.1S-luc were stained with PE Mouse IgG2a, κ Isotype Ctrl (FC) Antibody (Biolegend, 400214) as negative control. Data were collected comprised of 10,000 cell events and 1,000 beads events. Analysis software is QuickCal v 3.0 software provided by Bangs Laboratories. The ABC value of BCMA (0 μg) is defined as ABC zero.
Immunoblotting
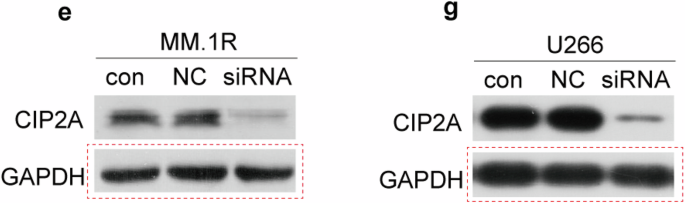
The Nb1, Nb2 and NbT CAR T cells were lysed with loading buffer (Sangon Biotech C516031) with or without dithiothreitol (DTT) followed by boiling. The protein samples were separated on the 8% gel (Smart-Lifesciences SLE021) using electrophoresis and then electro-transferred onto PVDF membranes (Millipore). The membranes were blocked in a blocking buffer (5% no-fat milk in phosphate-buffered saline with Tween 20) and incubated with primary antibody overnight at 4 °C, and then an appropriate Horseradish peroxidase (HRP)-conjugated secondary antibody. The primary antibody used was MonoRabTM Rabbit Anti-Camelid VHH Antibody, mAb (GenScript A01860; 1:1000 dilution). Protein expression was detected by enhanced chemiluminescence (EpiZyme SQ201). The stripping buffer (Beyotime Biotechnology P0025) was used to strip and re-probe western blot membranes. Then the membrane was blocked and incubated with GAPDH antibody (Beyotime Biotechnology AF1186; 1:2000 dilution) for detection.
Statistical analysis
Experiments were carried out in triplicate or quadruplicate as indicated in the corresponding figure legends. For cytotoxicity assays of multiple myeloma cell lines (Fig. 6a and Supplementary Fig. 6a), cytokine releasement (Fig. 6b), and immune synapse F-actin intensity (Fig. 6d), unpaired two-tailed Student’s t-test was performed on each grouped sample without adjustments for multiple comparisons. For all other experiments, the data were assessed for normality and equality of variances using the Shapiro-Wilk test and Brown-Forsythe test, respectively. For normally distributed data with equal variance, the one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test was used to assess the differences among different groups. If the normality or equal variance conditions were violated, the Kruskal-Wallis test followed by Dunn’s multiple comparisons correction was used. Data are presented as mean ± SEM. All statistical analyses were performed using GraphPad Prism software v.9 or R.



Comments (0)