
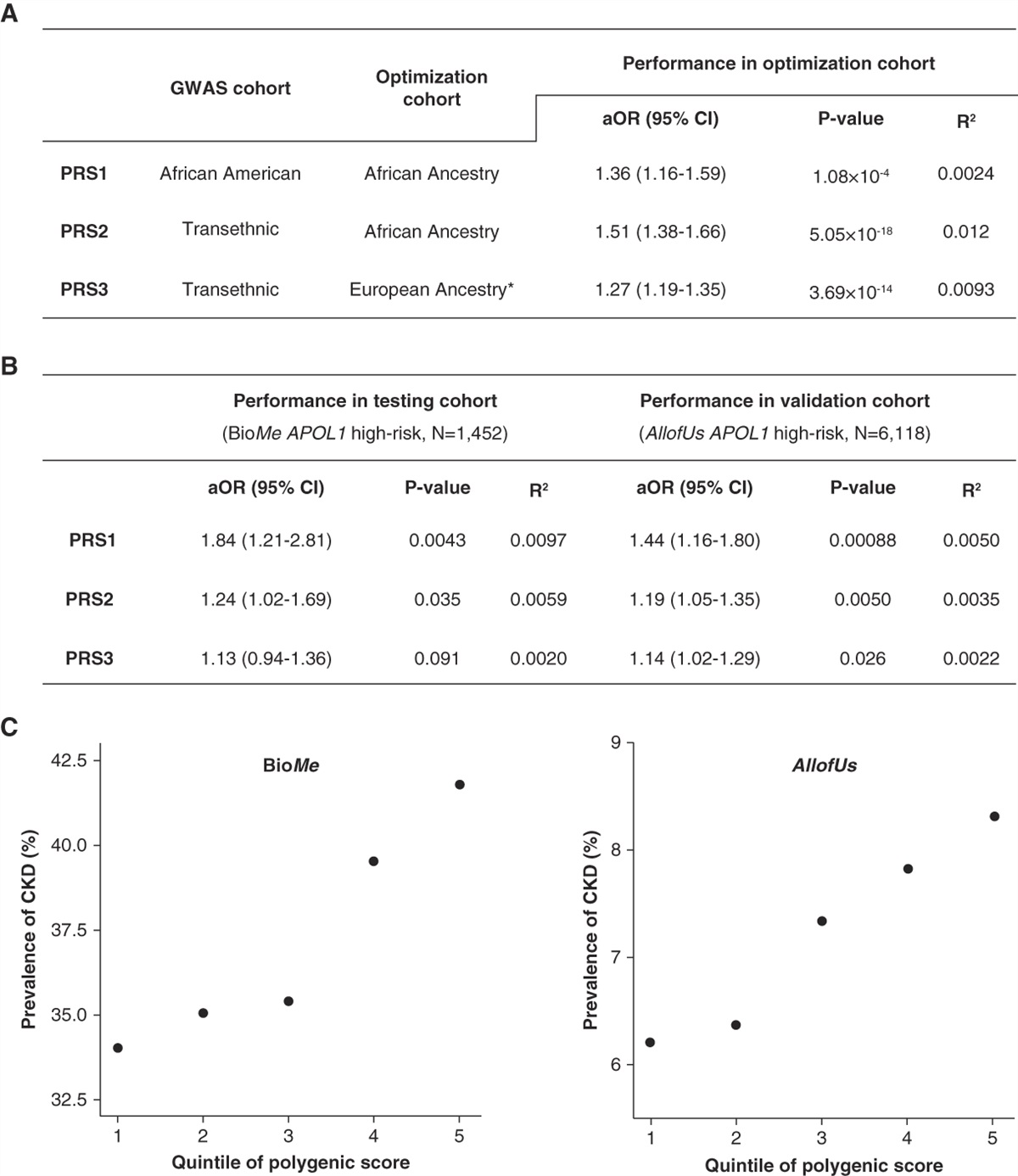
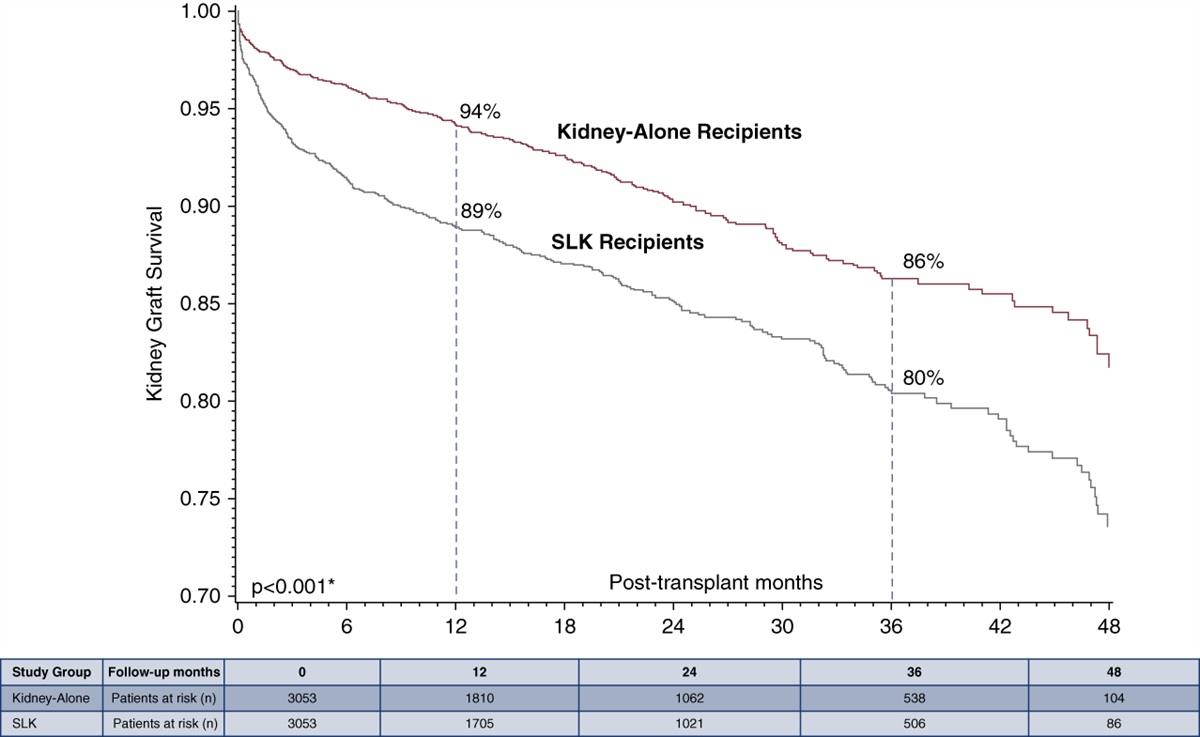
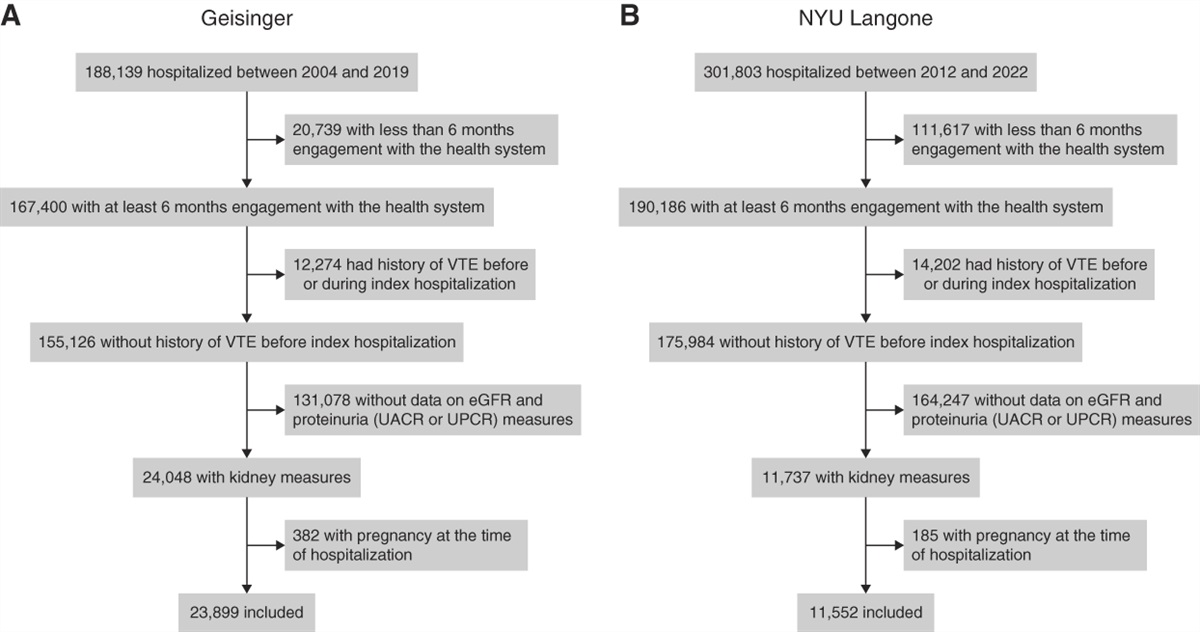
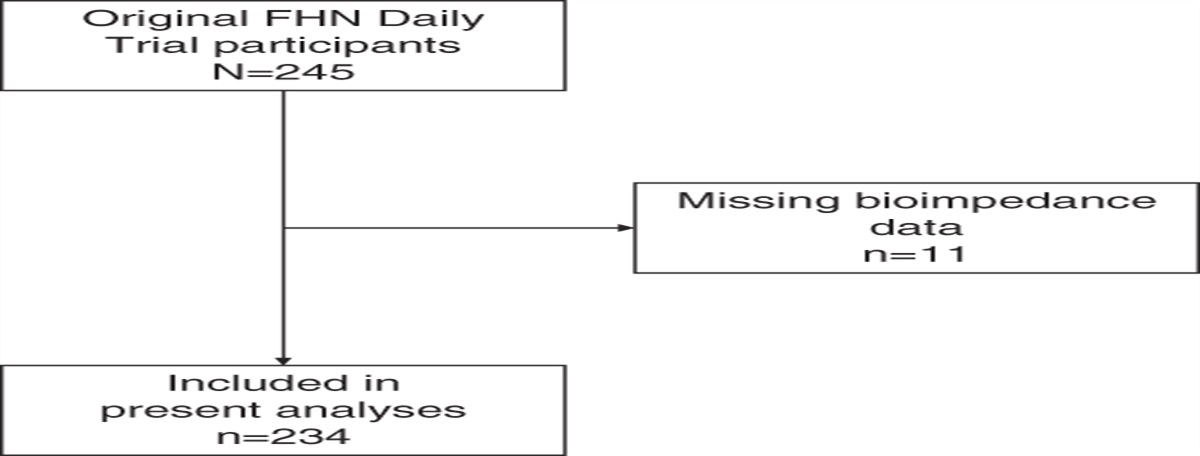
記住我

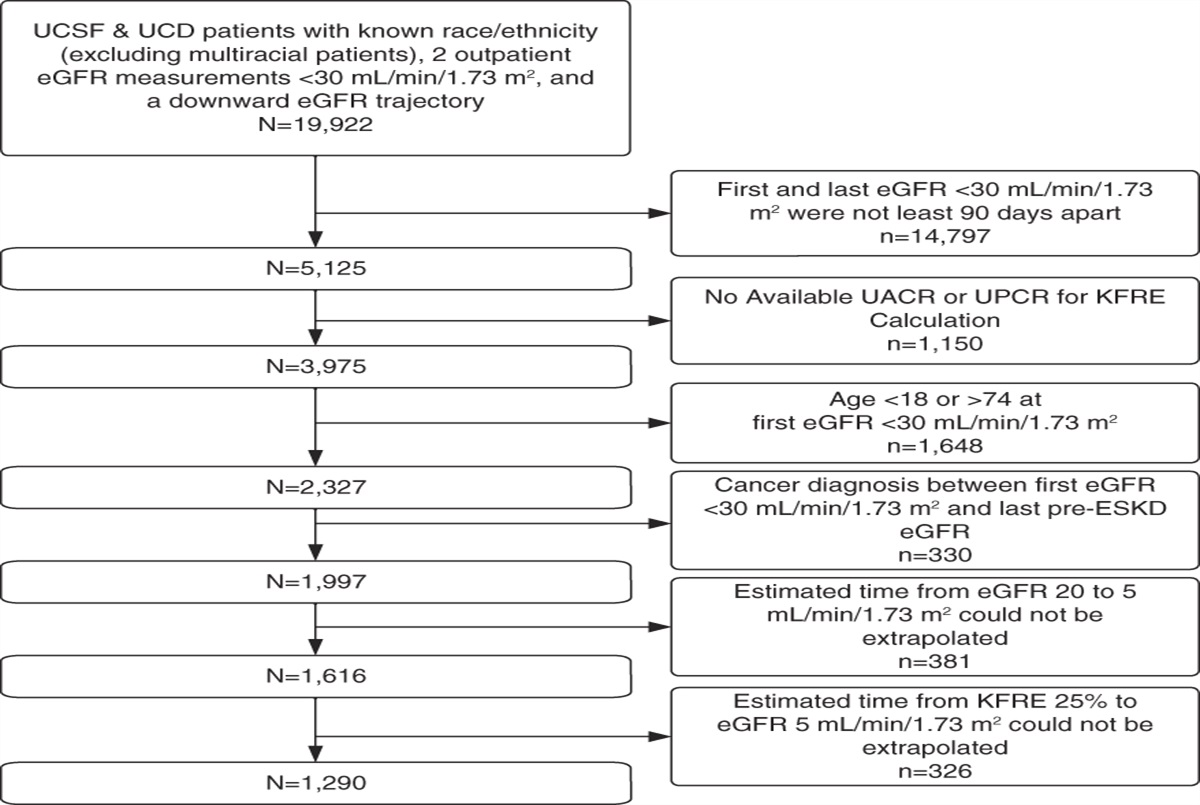
In the pursuit of BP-lowering therapies, few investigations have been as fruitful as efforts to thwart the renin-angiotensin-aldosterone system (RAAS).
A role for renin in BP regulation was first demonstrated in the late 19th century by the Finnish physiologists Tigerstedt and Bergman, who injected kidney extracts (labeled Renin) into rabbits and showed BP increases.1
Subsequently, as nicely summarized in a review article cataloging discovery in the RAAS,2 more than 30 years later, Harry Goldblatt demonstrated reproducible models of BP elevation using a clamp to reduce flow in the renal artery, showing that ischemia-provoked renin release mediated this increase in dogs. International efforts led to the discovery of a product after renin's enzymatic cleavage of the circulating protein angiotensinogen, dubbed angiotonin by researchers in Cleveland and hypertensin by researchers in Buenos Aires, Argentina. By mutual agreement, these names were merged into angiotensin. Active angiotensin-II (Ang-II) generation involved the conversion of angiotensin-I to the potent octapeptide Ang-II by endothelium-bound angiotensin-converting enzyme, which was noted to be particularly abundant in the lung.
Linking aldosterone into the RAAS came when studies showed that Ang-II not only raised arterial BP but also potentiated the release of electrocortin from the adrenal gland. Almost in parallel with renin system discoveries, the separation of glucose actions from electrolyte actions of adrenal cortex hormones, as nicely reviewed in the memoirs of James and Sylvia Tait,3 showed the existence of a hormone that regulated sodium and potassium transport, different from cortisone. The sodium retention from this hormone, aldosterone, whose release was significantly stimulated by Ang-II, provided a link between the vasoconstrictive and volume aspects of BP control.
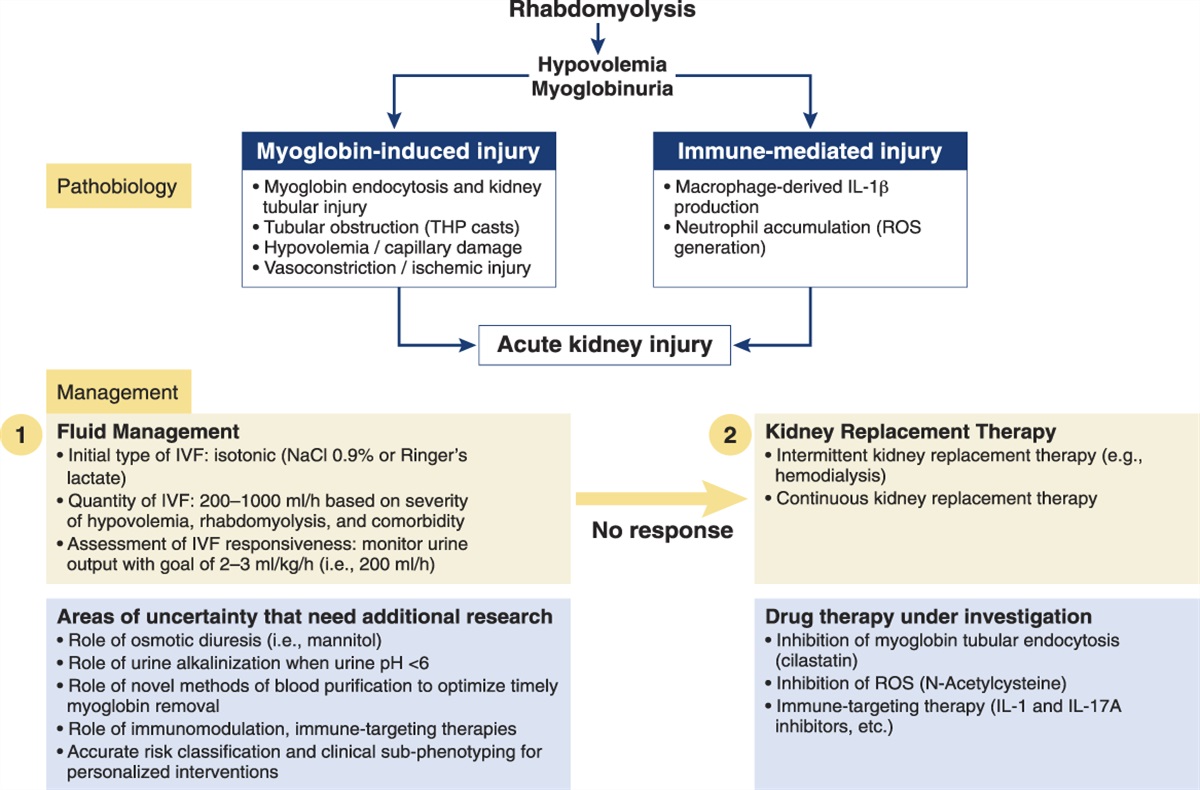
Aldosterone and BP RegulationA clinical role for aldosterone in hypertension emerged from a hypertension case presented by Conn in the 1950s, wherein removal of a 4-cm right adrenal tumor producing aldosterone resulted in a cure of hypertension in a 34-year-old woman.4
Aldosterone has subsequently been shown to be both a mediator of hypertension and a component of the pathophysiology attending comorbidities, such as CKDs and nephrosis, heart failure, and liver cirrhosis. A relative excess of aldosterone contributes to about 20% of treatment-resistant hypertension and is thought to participate in low-renin hypertension, which affects up to 25% of patients with essential hypertension.5
When aldosterone binds to the mineralocorticoid receptor, it stimulates the reabsorption of sodium by the distal kidney tubules and the colonic epithelium. The retention of sodium expands the circulating volume and contributes to an increase in BP by this volume expansion, often leading to a reduction in antihypertensive drug efficacy. For those involved in hypertension care, aldosterone's role in supporting BP elevation despite the use of multiple antihypertensive medications, including diuretics, encouraged the search for ways to mitigate aldosterone effects. Competition with aldosterone's binding to the mineralocorticoid receptor became possible when spironolactone was introduced (1959), furthered by eplerenone (2002) and finerenone (2021). These agents have proven to be effective in hypertension, heart failure, and in CKD progression.6 Moreover, there are mineralocorticoid receptor antagonists, such as esaxerenone, approved in other countries.
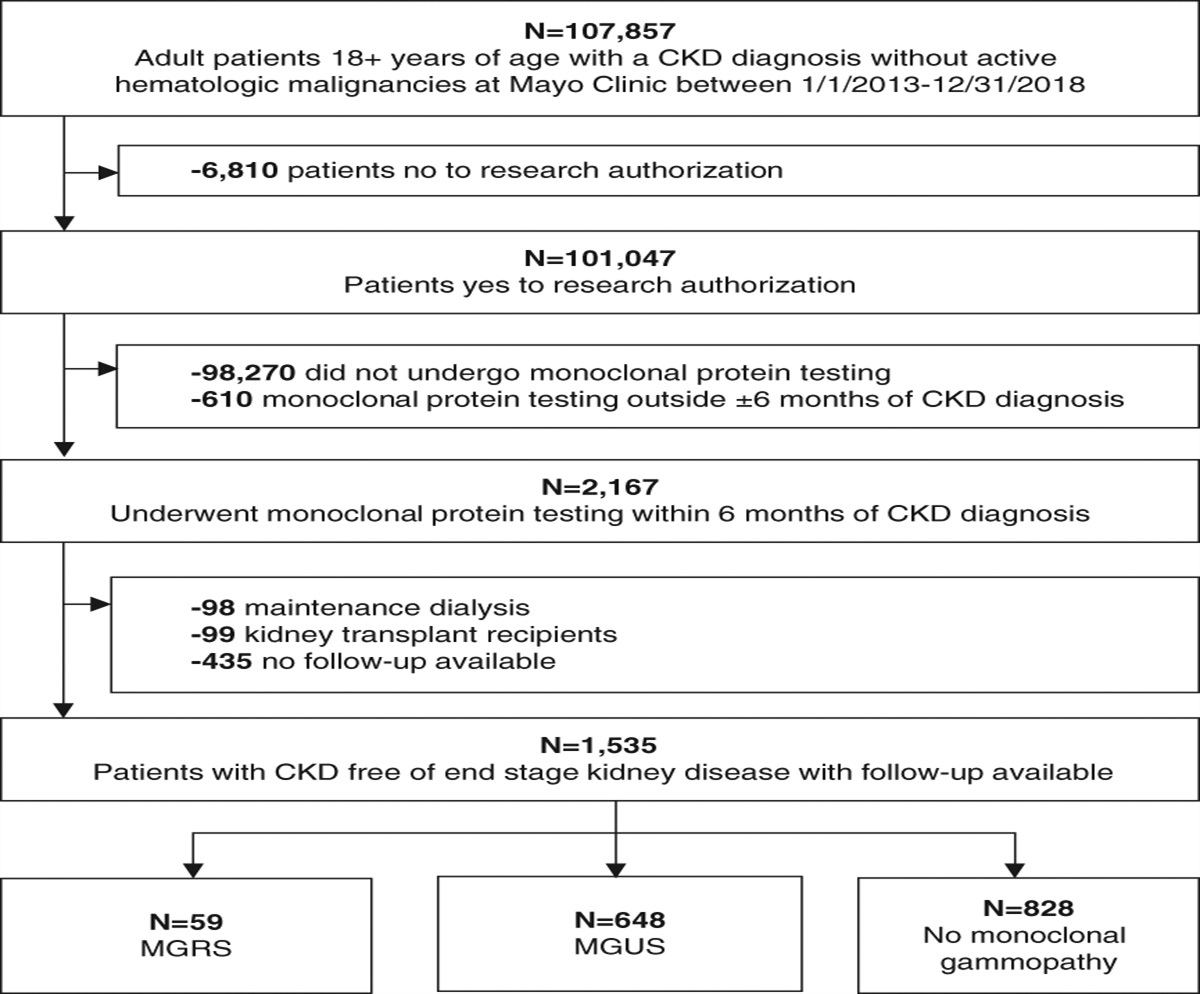
However, their efficacy comes at a price. The off-target endocrine effects, such as gynecomastia in men and amenorrhea in women with spironolactone, along with the potential for hyperkalemia with all three approved agents, are important clinical considerations. In addition, there are potential fibrotic and inflammatory actions mediated by aldosterone that occur independently of the receptor through aldosterone's binding to G-protein coupled receptors7 (Figure 1). Since mineralocorticoid receptor antagonists promote an increase in circulating aldosterone concentrations, the potential for increased actions of aldosterone independent of the mineralocorticoid receptor, along with the clinical challenges using them contributed enthusiasm to the idea of blocking aldosterone synthesis.
 Figure 1:
Figure 1: Aldosterone effects on BP. Shown are various aspects of aldosterone physiology, including synthesis in the z glomerulosa of the adrenal gland, BP effects mediated through sodium retention in the kidney, and other nongenomic actions attributed to aldosterone, such as inflammation and fibrosis on tissues like the heart, kidney, and blood vessel. Ang-II and ACTH stimulate aldosterone synthesis and release. Lower sodium intake and/or higher potassium intake also stimulate aldosterone synthesis and release. Aldosterone promotes UNa+ reduction and UK+ excretion, promoting higher BP, and it suppresses the release of renin from the kidney. ACTH, adrenal cortico tropic hormone; ALDO, aldosterone; Ang-II, angiotensin-II; CORT, corticosterone; UK+, urinary potassium; UNa+, urinary sodium; z, zona.
Aldosterone SynthesisA brief note on aldosterone synthesis will set the stage for the recently reported BrigHTN trial. The last steps in hormone synthesis for cortisol and aldosterone involve two enzymes that are about 93% homologous and whose genes sit next to one another on chromosome 8.8 Cortisol is produced by the action of cytochrome p450-11β-hydoxylase-1 on the precursor 11-deoxycortisol. In the aldosterone pathway, corticosterone is altered by the action of cytochrome p450-11β-hydroxylase-2 (aldosterone synthase) to produce aldosterone.
Although reports of aldosterone synthetic inhibition in men have appeared over the past few decades, progress toward their clinical usage has been slow. The difficulty with aldosterone synthetic blockade is the potential for simultaneous blockade of cortisol synthesis, found often with earlier agents.8 The emergence of compounds highly selective for aldosterone synthase while sparing cortisol production was the step forward for trials with compounds like baxdrostat.9
Aldosterone Synthesis InhibitionThe BrigHTN study was a four-arm, randomized, 12-week, double-blind trial in treatment-resistant hypertensive patients using three different doses of baxdrostat versus placebo in 275 patients, 248 of whom completed the10 study. Despite a substantial office systolic BP placebo response of 9 mm Hg, the 1-and 2-mg doses (the middle and highest doses of baxdrostat) showed significant additional systolic reductions of 8 and 11 mm Hg after 12 weeks of treatment, with an effect robust enough to prompt the Data Safety and Monitoring Board to recommended terminating the trial, following a planned interim analysis.
The investigators reported that the baxdrostat was well tolerated, with three occurrences of hyperkalemia (K+ >6.0 mEq/L) which responded to temporary-blinded drug withdrawal, dietary reinforcement, and reintroduction of study drug without recurrence in two instances. The third participant was withdrawn from the study because of unrelated urosepsis.
The definition of treatment resistance in BrigHTN was office BP 130–180/80–110 mm Hg on three drugs, including a diuretic. They excluded patients with an eGFR <45 ml/min per 1.73 m2, an important consideration when considering hyperkalemia occurrence. Enrolled participants had a body mass index of 32 kg/m2, and roughly 40% had diabetes, two comorbidities in which aldosterone is known to play a substantial role in BP elevation.
Not atypical in the journey of such compounds from clinical trials into clinical usage, the recently reported HALO trial (https://www.acc.org/latest-in-cardiology/clinical-trials/2023/03/01/23/34/halo accessed March 30, 2023) found a smaller and statistically nonsignificant reduction in office systolic BP, comparing the same three doses of baxdrostat with placebo in patients with uncontrolled hypertension on a RAS-blocking drug alone or as dual therapy with a diuretic or calcium channel blocker. In the HALO trial, a larger than expected placebo response of 16.6 mm Hg in systolic BP after 8 weeks lead to the nonsignificant reductions, raising possible protocol adherence and medication adherence issues, although all three doses of baxdrostat lowered circulating aldosterone concentrations. Publication of the HALO results should shed further light on these findings.
Next Steps?Exposure times longer than 12 weeks will be useful to monitor consequences of long-term aldosterone reduction, including breakthrough elevations in deoxycorticosterone. In addition, eGFR levels below the 45 ml/min per 1.73 m2 will need to be tested, as hyperkalemia is more likely with lower kidney function. Another question to address is a comparison of an aldosterone synthase inhibitor with a mineralocorticoid receptor antagonist given at full doses and possibly in conjunction with an antagonist. In the meantime, studies of aldosterone synthase inhibition with baxdrostat in patients with primary aldosterone excess (www.ClinicalTrials.govNCT04605549) and CKDs (www.ClinicalTrials.govNCT05432167) are underway, and results are awaited.
DisclosuresR.R. Townsend reports consultancy for BD, Cytel, IONIS, Medtronic, and Regeneron; research funding from NIH; and royalties from UpToDate.
FundingNone.
AcknowledgmentsThe content of this article reflects the personal experience and views of the author(s) and should not be considered medical advice or recommendation. The content does not reflect the views or opinions of the American Society of Nephrology (ASN) or CJASN. Responsibility for the information and views expressed herein lies entirely with the author(s).
Author ContributionsConceptualization: Raymond R. Townsend.
Writing – original draft: Raymond R. Townsend.
Writing – review & editing: Raymond R. Townsend.
References 1. Phillips MI, Schmidt-Ott KM. The discovery of renin 100 years ago. News Physiol Sci. 1999;14(6):271–274. doi:10.1152/physiologyonline.1999.14.6.271 2. Skrbic R, Igic R. Seven decades of angiotensin (1939-2009). Peptides. 2009;30(10):1945–1950. doi:10.1016/j.peptides.2009.07.003 3. Tait JF, Tait SA. A decade (or more) of electrocortin (aldosterone). Steroids. 1988;51(3-4):213–250. doi:10.1016/0039-128x(88)90016-5 4. Conn JW. Presidential address. I. Painting background. II. Primary aldosteronism, a new clinical syndrome. J Lab Clin Med. 1955;45(1):3–17. 5. Johnston JG, Welch AK, Cain BD, Sayeski PP, Gumz ML, Wingo CS. Aldosterone: renal action and physiological effects. Compr Physiol. 2023;13(2):4409–4491. doi:10.1002/cphy.c190043 6. Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386(10008):2059–2068. doi:10.1016/s0140-6736(15)00257-3 7. Williams JS. Evolving research in nongenomic actions of aldosterone. Curr Opin Endocrinol Diabetes Obes. 2013;20(3):198–203. doi:10.1097/med.0b013e328360c200 8. Azizi M, Amar L, Menard J. Aldosterone synthase inhibition in humans. Nephrol Dial Transplant. 2013;28(1):36–43. doi:10.1093/ndt/gfs388 9. Freeman MW, Bond M, Murphy B, Hui J, Isaacsohn J. Results from a phase 1, randomized, double-blind, multiple ascending dose study characterizing the pharmacokinetics and demonstrating the safety and selectivity of the aldosterone synthase inhibitor baxdrostat in healthy volunteers. Hypertens Res. 2023;46(1):108–118. doi:10.1038/s41440-022-01070-4 10. Freeman MW, Halvorsen YD, Marshall W, et al. Phase 2 trial of baxdrostat for treatment-resistant hypertension. N Engl J Med. 2023;388(5):395–405. doi:10.1056/nejmoa2213169
留言 (0)