Study design and participants
From April 2021 to August 2021, a total of 62 newly diagnosed T2D patients were consecutively enrolled at the Department of Endocrinology in Beijing Chao-yang Hospital Affiliated with Capital Medical University. All the patients had been diagnosed with T2D within the previous three months according to the 2020 American Diabetes Association diagnostic criteria, and met the following inclusion criteria: (1) aged 20–79 years old; (2) hemoglobin A1c (HbA1c) 7–10%. None of the patients had received anti-diabetic drugs before enrollment. None of the patients had any history of coronary artery disease, liver or renal function impairment, infectious disease, systemic inflammatory disease, hematological diseases, thyroid disease, autoimmune diseases, or cancer. Patients with ketoacidosis and hyperglycemic hyperosmolar status, and those who were pregnant or possibly pregnant, or ingesting agents influencing glucose or lipid metabolism were also excluded.
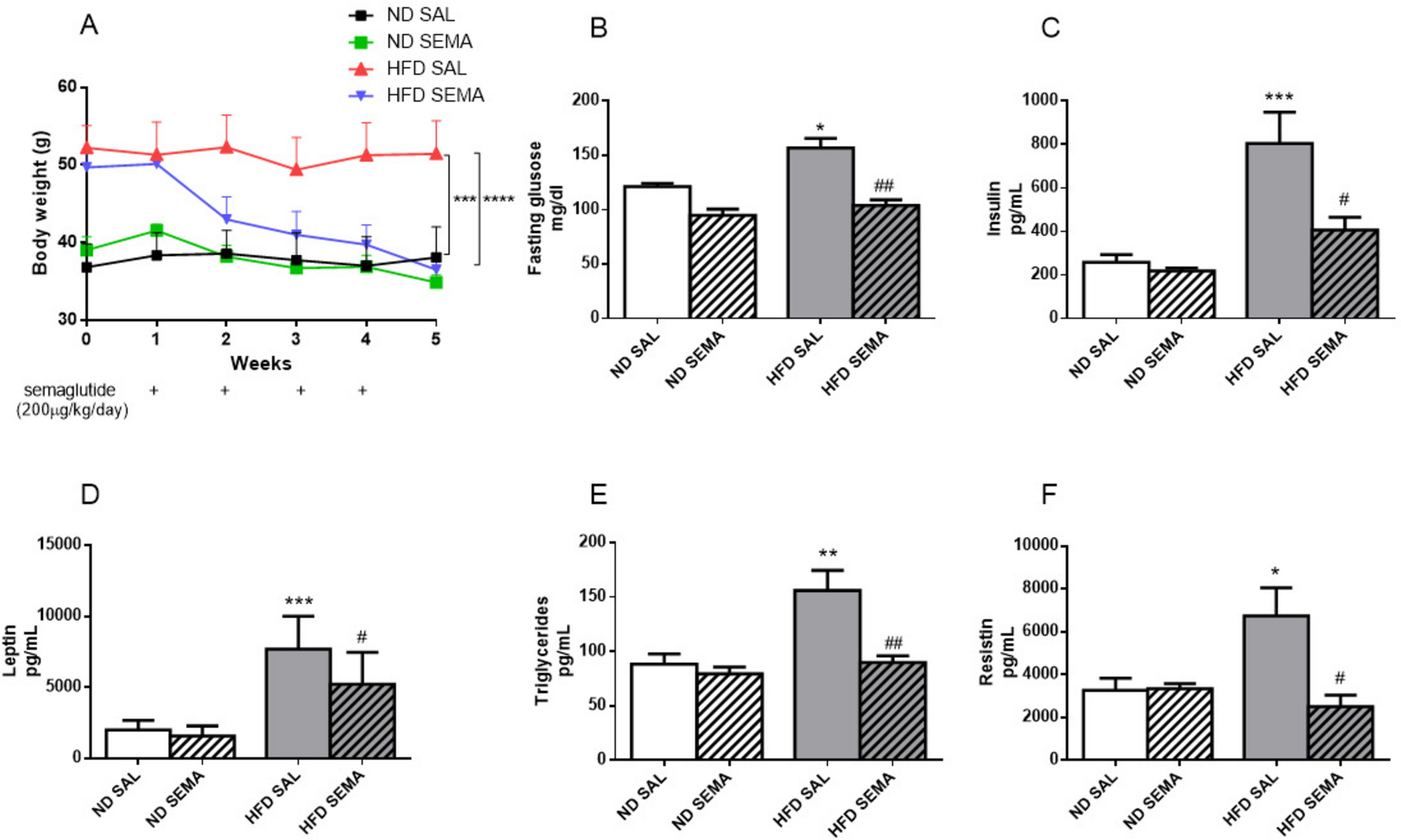
All the T2D patients received 12 weeks of dapagliflozin treatment (10 mg/d, AstraZeneca). During this time, the subjects received no other additional treatments. Patients were followed up every 4 weeks, and side effects were recorded at each visit. Three patients dropped out of the study due to urinary tract infections, and 2 patients dropped out due to self-discontinuation. During the 12 weeks of dapagliflozin treatment, hypoglycemia, ketoacidosis, or other side effects were not observed in any patients.
This study was conducted according to the principles of the Declaration of Helsinki, and approved by the Ethics Committee of Beijing Chao-yang Hospital Affiliated with Capital Medical University. Written informed consents were obtained from all participants.
Clinical and biochemical measurements
The information about health status and medications was collected by two skilled nurses using a standard questionnaire. Clinical and biochemical measurements were performed at baseline and after 12 weeks of dapagliflozin treatment. Height and weight were measured to the nearest 0.1 cm and 0.1 kg by the same trained group, respectively. BMI was calculated as the weight in kilograms divided by the height in meters squared. Fasting venous blood was collected in the morning after an overnight fast. Biochemical parameters were measured immediately and serum was stored at − 80 °C for proteomic and metabolomic analysis after centrifugation at 1500g for 20 min at 4 °C.
Serum triglyceride (TG) was measured by a glycerol lipase oxidase reaction, total cholesterol (TC) by an enzymatic cholesterol oxidase reaction, and high-density lipoprotein cholesterol (HDL-C) and low-density lipoprotein cholesterol (LDL-C) by a direct assay using an autoanalyzer (Hitachi 747, Roche Diagnostics, Germany). Nonesterified fatty acid (NEFA) concentrations were determined by enzymatic colorimetric assays (Hitachi 747, Roche Diagnostics, Germany). Fasting blood glucose (FBG) was detected using the glucose oxidase method (Hitachi 747, Roche Diagnostics, Germany). Fasting plasma insulin (FINS) was determined by the chemiluminescence method (Dimension Vista, Siemens Healthcare Diagnostics, Germany). HbA1c was estimated by high-performance liquid chromatography using the HLC-723G7 analyzer (Tosoh Corporation, Tokyo, Japan). Serum creatinine (CREA) level was measured by the picric acid method (Hitachi 747, Roche Diagnostics, Germany). Homeostasis model assessment of insulin resistance (HOMA-IR) was calculated according to the following formula: HOMA-IR = FINS (mIU/L) × FBG (mmol/L)/22.5 [12]. The estimated glomerular filtration rate (eGFR) was calculated using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation [13].
ProteomicsChemicals and reagents
All chemicals used were mass spectrometry (MS) grade or higher. Methanol, acetonitrile (ACN), and formic acid (FA) were purchased from Fisher Scientific (Thermo Fisher Scientific, Inc. USA). Sodium dodecyl sulfate (SDS), ammonium bicarbonate (NH4HCO3), trifluoroacetic acid (TFA), DT-Dithiothreitol (DTT), iodoacetamide (IAA), lysyl endopeptidase (LysC), urea, and trypsin were obtained from Sigma-Aldrich (St. Louis, USA).
Sample preparation
Total protein concentration was measured by a BCA Protein Assay Kit (Thermo Fisher Scientific, Inc. USA). 10 μL aliquot of serum samples (~ 600 μg proteins) were transferred into High Select™ depletion spin columns to deplete the top 14 abundant blood proteins following the manufacturer’s instruction (Thermo Fisher Scientific, Inc. USA). Then, 320 μL samples were centrifugated at 12,000g for 10 min in a 10kD ultrafiltration tube (Millipore, Burlington, MA, USA). 200 μL of 8 M urea was added to the sample and centrifuged at 12,000g for 10 min twice, and finally, 200 μL of 8 M urea was added. DTT solution was added to the sample until reaching final a concentration of 10 mM, and incubated at 37 ℃ for 30 min. IAA solution was added to a final concentration of 20 mM, incubated away from light at room temperature for 30 min, and centrifugated at 12,000g for 10 min. 200 μL of 50 mM NH4HCO3 solution was added to the sample and centrifuged at 12,000g for 10 min twice, and finally, 200 μL of 50 mM NH4HCO3 solution was added. Next, 4 μg of LysC (50:1, w/w, protein: enzyme) was added to each sample for incubation at 37 ℃ for 2 h, after which 4 μg trypsin (50:1, w/w, protein: enzyme) was added to the sample for incubation at 37 ℃ overnight. Finally, a final concentration of 1% TFA was added to terminate the reaction. Next, 100 μL of methanol was centrifuged at 600g for 1 min in SoLAμ HRP plate (Thermo Fisher Scientific, Inc. USA), after which 100 μL of 80% ACN 0.1% TFA and 200 μL of 0.1% TFA were added for centrifuging at 1000g for 1 min, respectively. Then, the sample was loaded to the SoLAμ HRP plate for centrifuging at 1000g for 2 min twice. Next, 200 μL 0.1% TFA was added to the sample for centrifuging at 1000g for 2 min, after which 100 μL 80% ACN 0.1% TFA was added for centrifuging at 1000g for 3 min. Then the eluate was dried at 40 ℃ using a Centrifugal concentrator. Next, the digested and desalted peptides were dissolved to 0.5 μg/μL with 0.1% FA, after peptide assay by using NanoDrop microvolume spectrophotometer (Thermo Fisher Scientific, Inc. USA), and 2 μg was loaded into liquid chromatography-mass spectrometry (LC–MS) for data-independent acquisition (DIA) analysis.
LC–MS analysis
DIA proteomic analysis was performed by LC–MS analysis using a UltiMate™ 3000 RSLC nano-LC system (Thermo Fisher Scientific, Inc. USA) with Q Exactive HFX™ quadrupole-electrostatic field orbitrap high resolution mass spectrometry (Thermo Fisher Scientific, Inc. USA). XCalibur 4.3 (Thermo Fisher Scientific, Inc. USA) was used for data acquisition. All samples were allocated in a random order, and technicians were blinded to the status of the samples. Quality control (QC) samples (pooled samples from equal aliquots of each sample) were used to monitor the MS performance. Further details are provided in the Supporting Information section.
The digested peptide was loaded onto the Trap Column (Acclaim PepMap C18, 3 μm, 100 Å, 75 μm*2 cm, Thermo Fisher Scientific, Inc. USA) with buffer A (0.1% FA), and subsequently separated on the analytical column (Acclaim PepMap C18, 2 μm, 100 Å, 75 μm*25 cm, Thermo Fisher Scientific, Inc. USA). The trap column was eluted with different gradients of buffer B (0.1% FA, 80% ACN). The gradient of buffer B was from 3 to 6% in 3 min, 8% to 30% in 95 min, 30% to 99% in 4 min, and 99% to 99% in 5 min. The column flow rate was maintained at 300 nL/min.
A mass spectrometer with electrospray at an inlet voltage of 2.1 kV was used. The temperature of the heated capillary was set at 300 °C. After ionization, MS1 was performed using an Orbitrap Fusion Lumos (Thermo Fisher Scientific, Inc. USA). Fragmentation was achieved by high-energy collisional dissociation (HCD) with a collision energy of 32%. Data were obtained in DIA mode.
Data processing
DIA data were processed using DIA-NN 1.8 (The Francis Crick Institute, UK). A previously generated human chromatography library was used in the targeted analysis of DIA data against the human reference proteomics database. Default settings were used unless otherwise noted. Protein identifications were accepted if the false discovery rate (FDR) < 1% by the Scaffold Local FDR algorithm. When samples failed quality control, proteins were removed. Proteomic datasets were filtered for 75% valid values across all samples (proteins with > 25% missing values were excluded from downstream statistical analysis). Then, the K-nearest algorithm (sample-wise) was employed to impute the missing values. Protein intensities were then log-transformed for further statistical and bioinformatics analysis.
metabolomicsChemicals and reagents
All chemicals used were MS grade or higher. Methanol, ACN, and FA were purchased from Fisher Scientific (Thermo Fisher Scientific, Inc. USA). Ammonium acetate was purchased from Sigma-Aldrich (St. Louis, USA). Isotope labeling internal standards were purchased from Cambridge Isotope Laboratories (Tewksbury, MA, USA) and Toronto Research Chemicals (Toronto, Canada). Ultra-pure water (18.2 MΩ·cm) was prepared using a Milli-Q purified water system (Merck KGaA, Darmstadt, Germany).
Sample preparation
After thawing at 4 ℃, 120 μL of samples were transferred to a 96-well plate (Thermo Fisher Scientific, Inc. USA), and 480 μL methanol-ACN extract (containing isotope labeled internal standards: Taurine-d4 1.0 μg/mL, Hippuric acid-d5 1.0 μg/mL, Chlorophenylalanine 1.0 μg/mL, Acylcarnitine(12:0)-d9 0.2 μg/mL, Acylcarnitine(18:0)-d3 0.2 μg/mL, Palmitic acid-13C16 0.2 μg/mL, Stearic acid-d35 1.0 μg/mL, Chenodeoxycholic acid-d4 1.0 μg/mL) was added for vortex oscillation for 5 min. After centrifugation at 2000g for 20 min at 4 ℃, two 200 μL aliquots of each extract were transferred to another 96-well plate. The QC sample was prepared by mixing an equal aliquot of the supernatants from all samples. The extracts were concentrated and dried by decompressed centrifugation (Labconco Corporation, Kansas City, USA). After adding 80 μL polar complex solution, the supernatant of extracts was collected and transferred to a 96-well plate for further metabolomic analysis.
Ultra-high performance liquid chromatography-high resolution mass spectrometry analysis
The specific technical detection method was consistent with the study by Du et al. [14]. Non-targeted metabolomics analysis was conducted using a Ultimate™ 3000 ultra-high performance liquid chromatography coupled with Q Exactive™ quadrupole-Orbitrap high resolution mass spectrometer system (Thermo Fisher Scientific, Inc. USA). The hydrophilic fraction of metabolite extract was injected into the analytic workflow randomly. Technicians were blinded to the status of samples. Further details were provided in the Supporting Information section. All the data was acquired in profile format.
Data processing
Compound Discoverer software (Thermo Scientific, San Jose, USA) was used for comprehensive component extraction. The hydrophilic metabolites were structurally annotated by searching acquired MS2, local high-resolution MS/MS spectrum libraries, as well as mzCloud library (Thermo Scientific, San Jose, USA). Besides, the exact m/z of MS1 spectra was searched via a local HMDB metabolite chemical database. Mass accuracy of precursor within ± 5 ppm was the prerequisite, and a fit score of relative isotopic abundance pattern > 70% was employed to determine the chemical formula. Furthermore, retention time as well as high resolution MS/MS spectra similarity was employed to strictly confirm the structural annotation of metabolites. The area under curve value extracted by XCalibur Quan Browser information was used as the quantitative information of metabolites, and all peak areas data for the annotated metabolites were exported into Excel software for trim and organization before statistics. Finally, the chemical identification results were annotated with classification criteria proposed by MSI (metabolomics standardization initiative). The metabolomic data from the two measurements were merged and trimmed for further data process. MetaboAnalyst 4.0 (www.metaboanalyst.ca) was used to filter missing values by the following criterion: the metabolites with features > 50% missing values. The remaining missing values were replaced by half of the minimum positive value in the original data. The metabolomics data were then log-transformed for further statistical and bioinformatics analysis.
Statistical and bioinformatical analysis
Differences in clinical parameters were analyzed using SPSS 22.0 (SPSS, Chicago, IL, USA). The distribution of continuous data was evaluated using Kolmogorov‐Smirnov test. For normally distributed data, continuous data were expressed as mean ± standard deviation. Because following the skewed distribution, the values of TG, FINS, and HOMA-IR were given as medians and upper and lower quartiles. Changes in parameters from baseline values within a group were evaluated using a paired t-test. Statistical significance was considered with two-tailed analyses as p < 0.05.
Principal component analysis (PCA) and orthogonal partial least square discriminant analysis (OPLS-DA) of proteins or metabolites were performed in SIMCA software (Umetrics AB, Umea, Sweden). A validation plot was used to assess the validity of the OPLS-DA model using permutation tests (n = 999). Differences of proteins or metabolites in baseline and after the treatment were analyzed by the Wilcoxon signed rank test for paired comparisons in the R statistical environment, version 4.1.3. Differentially abundant proteins or metabolites were identified by meeting the following criteria: (1) |log2 fold change (FC)|> 0.1375; and (2) the p value after the FDR multiple test correction (q value) < 0.05 by Benjamini–Hochberg method. Data visualization was conducted using R Studio [15, 16]. Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were conducted using the clusterProfiler package. Fisher’s exact test was used to evaluate the enrichment of the differentially abundant proteins, and a p value < 0.05 was considered significant. The correlations between differentially abundant proteins and metabolites were analyzed using Spearman rank correlation analysis, and heatmaps were drawn using R 4.1.3. The univariate receiver operating characteristic (ROC) curve by using the area under the curve (AUC) was applied to assess the accuracy of these changed metabolites and proteins to distinguish between the baseline and after dapagliflozin treatment.





Comments (0)